Somaya Alzeylae, Thowiba Awad, Elnazeer Hussien, Mosad Odah, Ashraf Ewis* and Mohamed Elhefny
Osteogenesis imperfecta (OI) comprises a group of connective tissue disorders characterized by bone fragility and reduced bone mineralization density. The disorder is clinically and genetically heterogeneous. Here, we present a case of 12 years old boy who was presented to the pediatric emergency department of Al-Qunfudah general hospital at southwestern Saudi Arabia with multiple repeated fractures due to trivial trauma. He got his first fracture of the femur at the age of 6 years and within a short period, due to the multiple and repeated fractures that involved his long bones and vertebrae, he became handicapped, depending on a wheeled chair in his movement. Genetic testing confirmed that the boy has a homozygous pathogenic variant in the SERPINF1 gene, consistent with the genetic diagnosis of autosomal recessive OI type VI. Both parents showed the familial heterozygous pathogenic variant in the SERPINF1 gene confirming a carrier state. Here we are going to present the case and follow up its diagnosis by biochemical bone profile and genetic analysis. We conclude that OI-type VI is a rare, recessively inherited bone mineralization disorder that has no cure yet, but a challenging multidisciplinary management approach is warranted.
HTML PDFShare this article
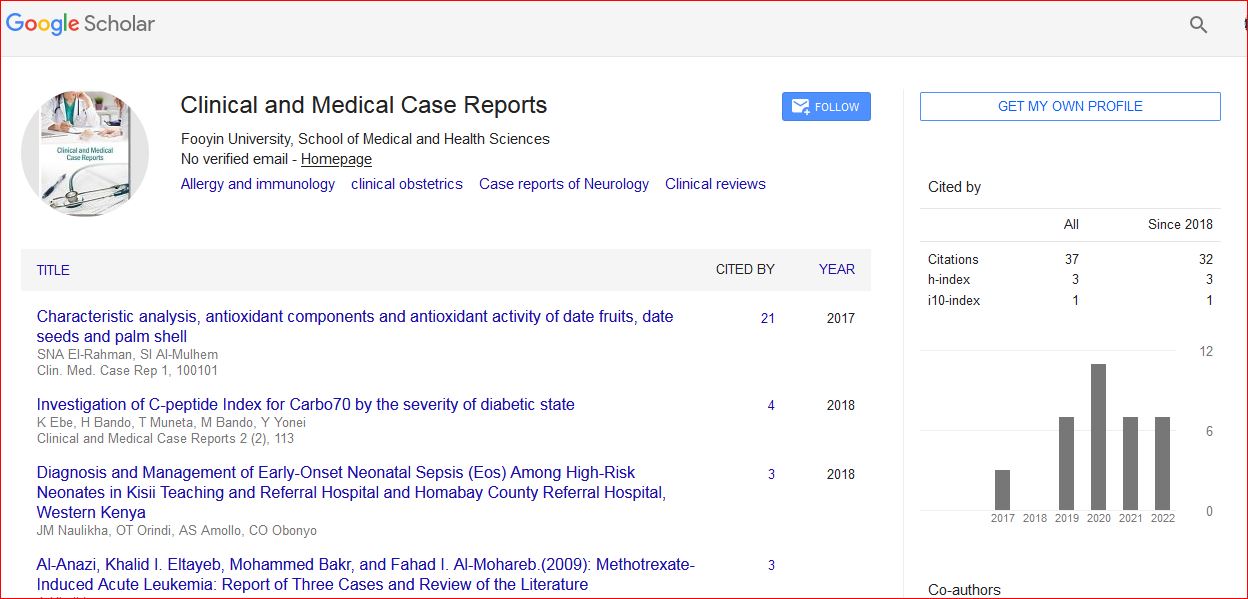
Clinical and Medical Case Reports received 53 citations as per Google Scholar report