Katja Kaastrup , Kirsten Grønbæk , Sine Reker Hadrup and Andreas Glenthøj
Aplastic anemia (AA) and paroxysmal nocturnal hemoglobinuria (PNH) are two rare and often concomitant hematologic diseases. Their development is - despite lack of decisive evidence - attributed autoimmune mechanisms. AA is characterized by cytopenias combined with a hypocellular bone marrow. The pathophysiology is believed to be immune-mediated with destruction of hematopoietic stem cells by autoreactive lymphocytes, a hypothesis supported by its response to immunosuppressive therapy. A large proportion of patients with AA also have PNH. In PNH, somatic mutations in the PIGA gene in hematopoietic stem cells blocks synthesis of the glycosylphosphatidylinositol (GPI) anchor. As a result, blood cells deriving from the GPI-deficient clone lack GPI-anchored proteins, most notably the complement inhibiting factors CD55 and CD59. The clinical manifestations primarily arise because of uncontrolled complement activation on erythrocytes and thrombocytes. This review summarizes current knowledge and theories about of the pathophysiology of both diseases with focus on immune mechanisms attributing to the two diseases.
PDFShare this article
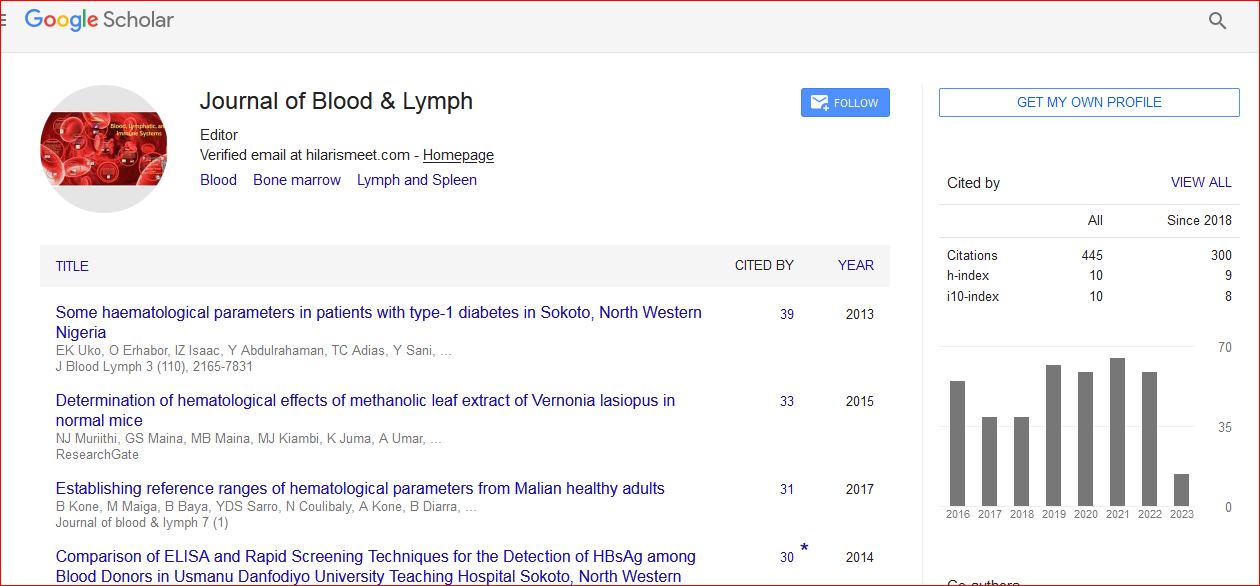
Journal of Blood & Lymph received 443 citations as per Google Scholar report