Alaa S. Shraim
The c-KIT is a transmembrane receptor tyrosine kinase that plays a crucial role in cancer progression. Deregulation of c-KIT has been detected in several human cancers. In particular, the two point mutations D816V and D816H in the kinase domains which induce ligand-independent activation of the kinase activity and acquired drug resistance. Therefore, c-KIT represents an attractive target for cancer therapy. Aptamers are emerging as a new promising and innovative class of nucleic acid therapeutic agents. In our study, a conventional SELEX approach was applied against the kinase domain of a group of c-KIT proteins (c-KIT WT, c-KIT D816V, and c-KIT D816H) to select RNA aptamers from a random RNA pool that can bind to the kinase domain of each c-KIT protein with high affinity and can selectively interfere with their kinase activities. Interestingly, our experimental data indicated that one of the candidate aptamers, called V15, can potently and specifically inhibit the in vitro kinase activity of mutant c-KIT D816V with an IC50 value that is 9 fold more potent than the FDA approved sunitinib drug tested under the same experimental conditions. Kinetic analysis revealed that V15 aptamer inhibits the kinase activity of the tested c-KIT kinases in a non-competitive manner. Another aptamer, named as H5/V36, showed the potential to functionally distinguish between the c-KIT WT, c-KIT D816H, and c-KIT D816V kinases by modulating the phosphorylation activity of each in a distinct mechanism of action and in a different potency. Finally, H5/V36 aptamer also revealed significant selectivity, since almost no inhibitory activity was detected after testing against two closely related kinases: PDGFRβ and JNK3 kinases. Taken together, our results suggest that these RNA aptamers may serve as a platform for the future development of novel aptamer-based targeted therapies for cancer.
PDFShare this article
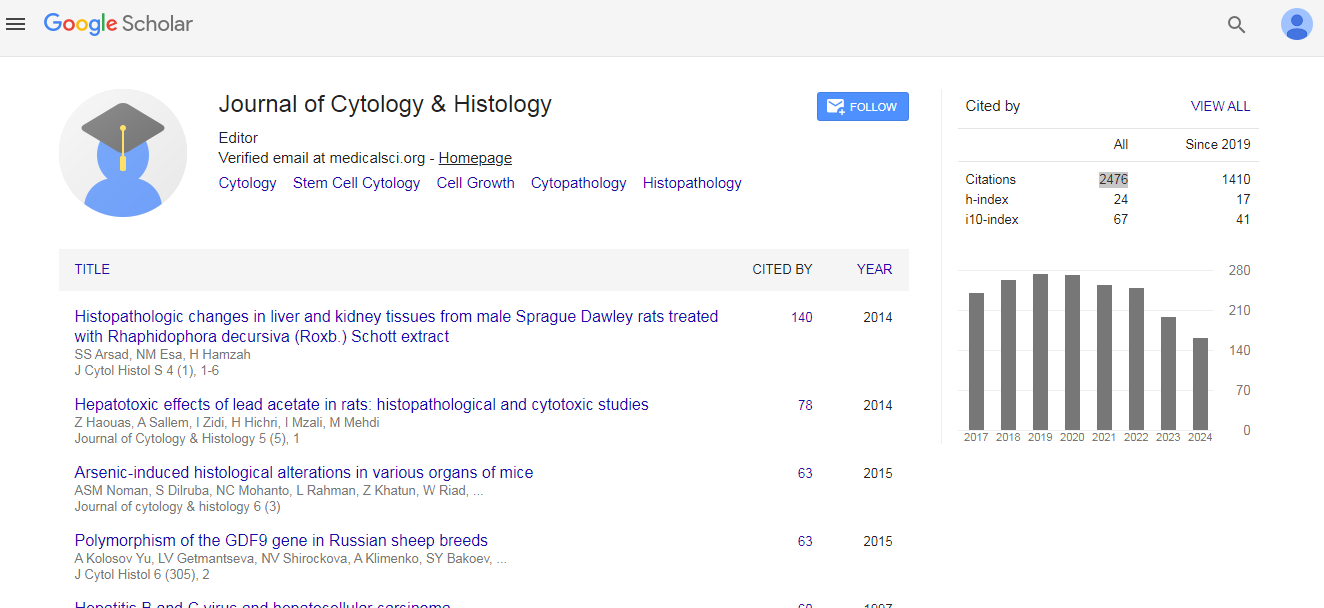
Journal of Cytology & Histology received 2476 citations as per Google Scholar report