Tal Thomas Sadeh
Cav1.4 is a retina-specific voltage-dependent Ca2+-channel that plays a regulatory role in sensory neurotransmission. Mutations in CACNA1F, encoding the conductive α1F subunit of Cav1.4, cause distinct eye dystrophies, including congenital stationary night blindness (CSNB), cone-rod dystrophy, and Åland eye disease. CANCA1F mutations detected in CSNB patients may be casual for the disease, however, the lack of functional validation prevents the provision of a diagnosis, and therefore, novel therapeutic targets. We have devised a protein-specific model that can predict the pathogenicity of these mutations that needs functional validation.
Membrane proteins like Cav1.4 are translocated from the endoplasmic reticulum to the plasma membrane, within Golgi vesicles. However, missense mutations may cause protein misfolding events that can reduce the level of expression, mislocalisation, and decrease the function. The misfolding of mutant proteins can be rescued by small molecules, such as chemical chaperones that stabilise protein folds and reduce non-native interactions, or proteostasis regulators that enhance protein folding and trafficking. Both classes of molecules can protect mutant proteins from degradation.
This suggests that small molecules have great potential as a valuable therapeutic approach for treating retinal, and other, protein misfolding diseases. I will use CSNB as an exemplar of this in this project to test the pathogenicity of novel CSNB variants of unknown significance identified in Manchester Centre for Genetic Medicine NHS diagnostic laboratory. These variants will validate our inhouse prediction tool and test the effect of small molecules on protein expression, localisation, and function.
Share this article
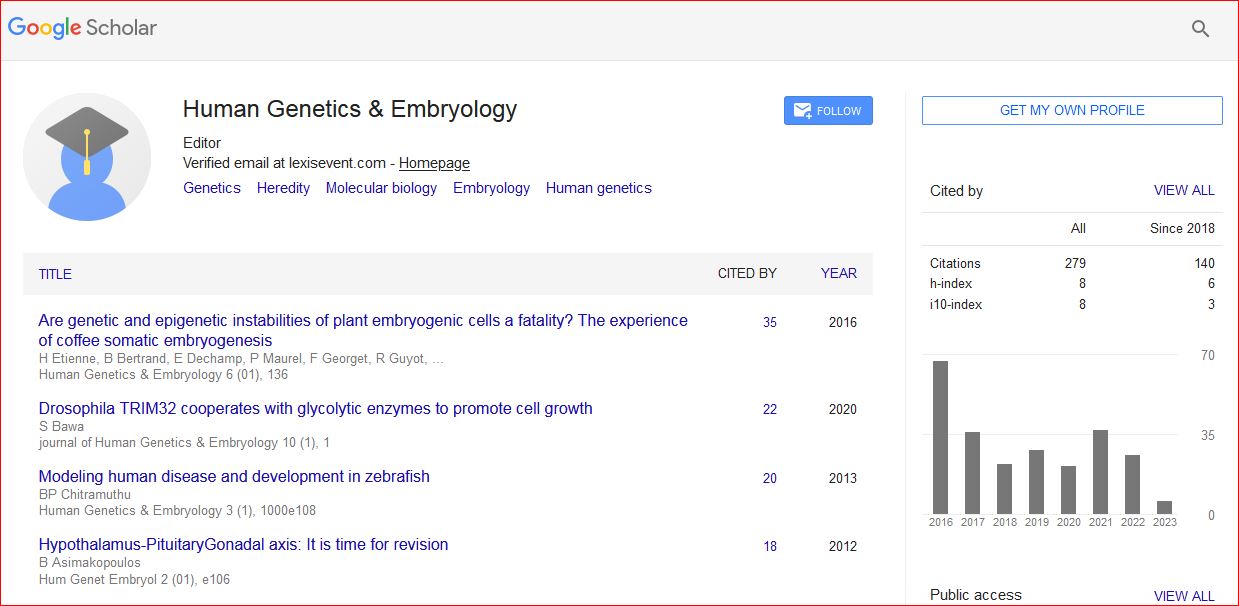
Human Genetics & Embryology received 309 citations as per Google Scholar report