Ioana Vasiliu, Bogdan Hancearuc, Dan Iliescu, Cipriana ?tef?nescu, Radu Popa, Delia Ciobanu, Leti?ia Leu?tean, Voichita Mogo? and Carmen Vulpoi
Pheochromocytoma and paraganglioma are catecholamine secreting tumors. Malignancy is uncommon - approximately 10% for pheochromocytoma and 20% for paraganglyoma. Surgery, when possible, is the first line treatment. Prognosis is poor because of a frequent local recurrence and/or metastases and the lack of specific chemotherapeutic agents. We report the case of a 60 years old man who was hospitalized at the age of 48 for episodes of paroxystic hypertension with spells. The high levels of vanillylmandelic acid (VMA), more than 50 mg/24h at 3 determinations, confirmed the excess of catecholamine, but the CT scan failed to reveal the tumor. The iodine-131- meta-iodobenzylguanidine (I-MIBG) scintigraphy showed the presence of a 1.5 cm nodule in the left abdominal paraaortic region. The patient refused surgery and had a satisfactory evolution with antihypertensive therapy. 11 years later he was admitted in the Vascular Surgery Department for acute ischemia of inferior limbs; a voluminous para-aortic tumor was diagnosed and resected. The pathology confirmed paraganglioma and described some atypical cells without being able to discriminate between benign or malignant pattern. Anamnesis could not identify any other case in the family. After another year he was admitted in the Endocrine Department for the reappearance of the adrenergic syndrome, with VMA at 30.8 mg/24h. The thoracic and abdominal CT-scan showed abdominal and thoracic metastatic tumors. The patient was referred to a specialized center were octeotride scintigraphy confirmed local recurrence and metastatic tumors in the lungs. He was treated with Sunitinib with a good initial response and he died after 16 years from the initial diagnostic of paraganglioma.
PDFShare this article
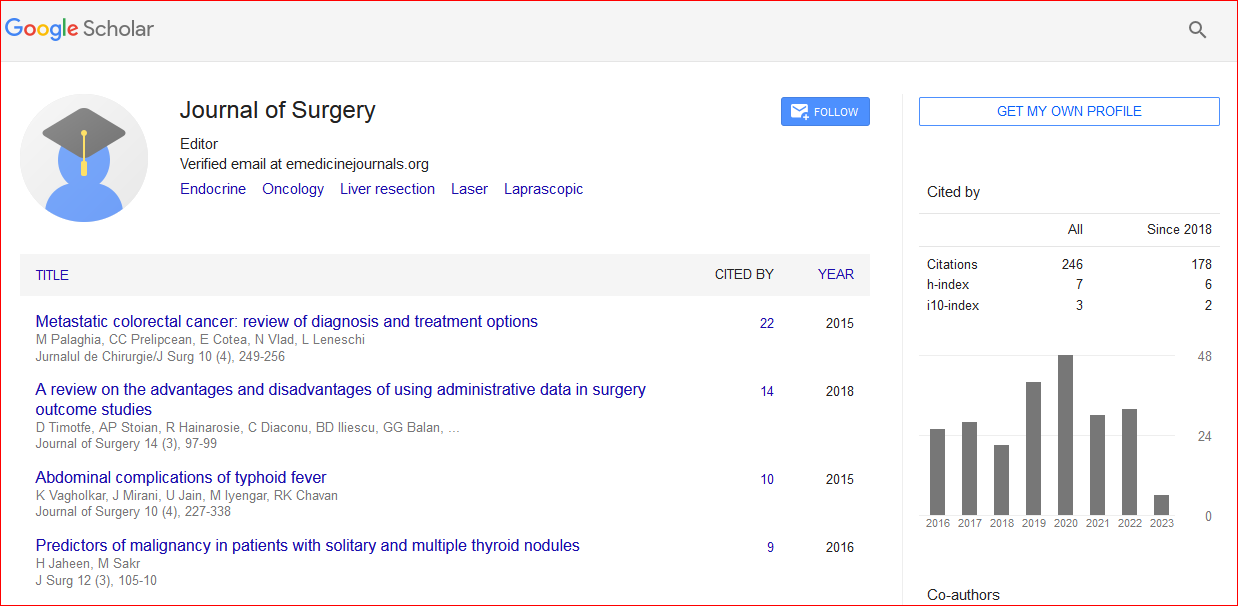
Journal of Surgery received 288 citations as per Google Scholar report