Shein-Chung Chow and Mo Liu
For approval of generic drug products, the United States Food and Drug Administration (FDA) has published several regulatory guidance to assist the sponsors in preparing documents, which provide substantial evidence for demonstration of bioequivalence between a generic (test) product and its innovative (reference) product (e.g., FDA, 1992, 2003) through the conduct of bioavailability and bioequivalence studies. Bioavailability and bioequivalence studies are usually conducted under crossover designs such as a standard 2x2 crossover design or a higher-order crossover design. Under a crossover design, bioequivalence is commonly evaluated using a two one-sided tests procedure (each at a 5% level of significance) or a 90% confidence interval approach. Bioequivalence is claimed if the constructed 90% confidence interval for the geometric mean ratio falls entirely within the bioequivalence limit of (80%, 125%). Statistical methods for bioequivalence evaluation are well established and widely accepted in the pharmaceutical industry since the publication of the FDA guidance in 2003. However, several practical issues are commonly encountered during the review of regulatory submissions of generic drug products. In this article, these issues are described. In addition, some recommendations for possible clarification and/or resolutions are made.
PDFShare this article
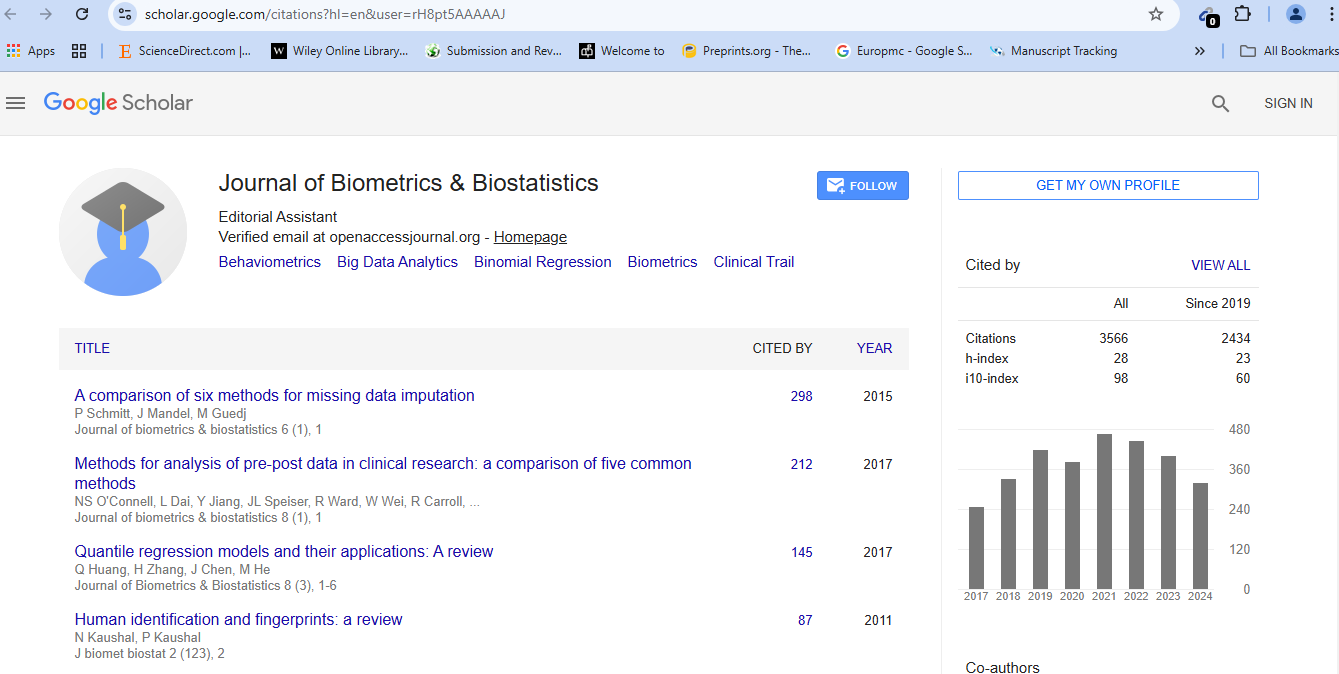
Journal of Biometrics & Biostatistics received 3496 citations as per Google Scholar report