Shea N. Gardner and Tom Slezak
With the flood of whole genome finished and draft microbial sequences, analysts need faster, more scalable bioinformatics tools for sequence comparison. An algorithm is described to find single nucleotide polymorphisms (SNPs) in whole genome data. It scales to hundreds of bacterial or viral genomes, and can be used for finished and/ or draft genomes available as unassembled contigs. The method is fast to compute, finding SNPs and building a SNP phylogeny in seconds to hours. It identified thousands of putative SNPs from all publicly available Filoviridae, Poxviridae, foot-and-mouth disease virus, Bacillus, and Escherichia coli genomes and plasmids. The SNP-based trees it generated were consistent with known taxonomy and trees determined in other studies. The approach described can handle as input hundreds of megabases of sequence in a single run. The algorithm kSNP is based on k-mer analysis using suffix arrays and requires no multiple sequence alignment.
PDFShare this article
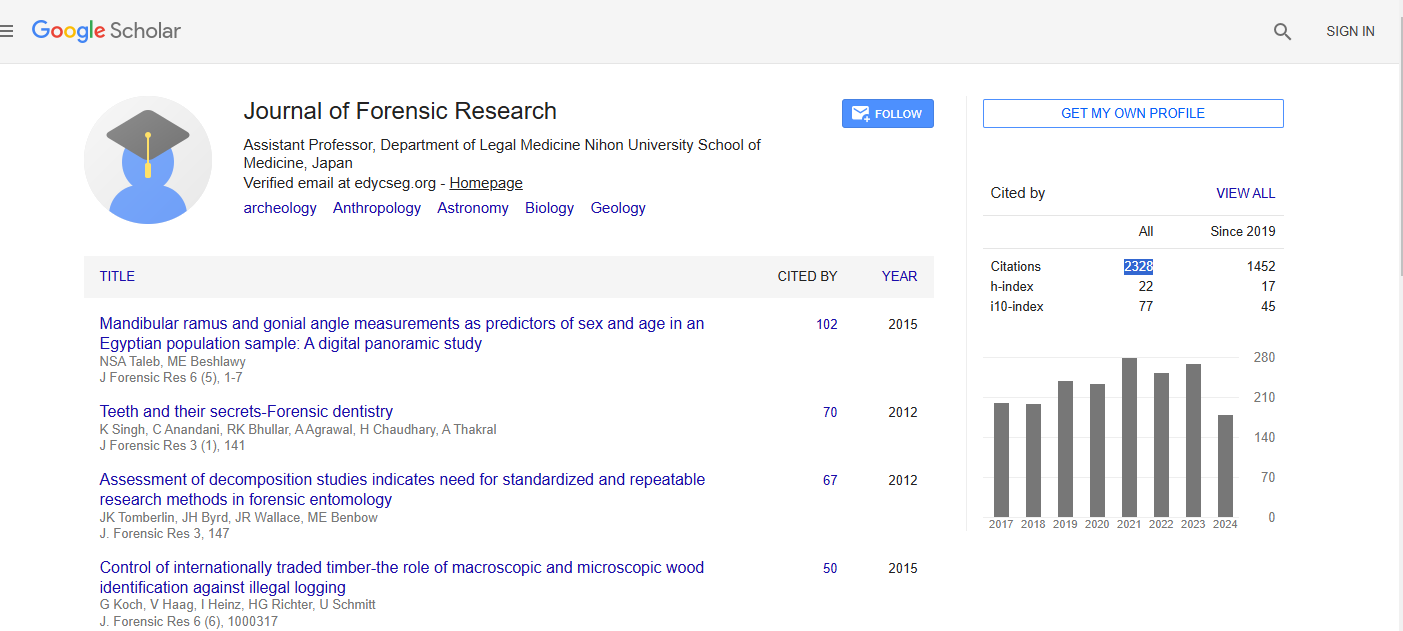
Journal of Forensic Research received 2328 citations as per Google Scholar report