Smigiel R, Kusmierska K, Pollak A, Szmyd K, Kosinska J, Polawski T, Kostrzewa G, Sasiadek MM and Ploski R
De Barsy syndrome is a rare autosomal recessive genetic disorder characterized by growth retardation, intellectual disability, a prematurely-aged appearance (progeroid features) and loose skin (cutis laxa) as well as eye abnormalities and others. Some cases of de Barsy syndrome have been linked with mutations PYCR1 or ALDH18A1. We describe a family with two siblings with clinically severe de Barsy syndrome in whom two novel mutations in ALDH18A1 (p.Glu100* and p.Arg724His) were found by clinical exome sequencing using TruSight One panel. The p.Glu100* is a novel mutation predicted to cause absence of the protein. The p.Arg724His has been found with low frequency (0.000016) but not in association with human disease; it has been scored as pathogenic by CADD, MetaSVM, Polyphen2, MutationAssessor, SIFT and MutationTaster. The level of ammonia in serum was determined in second sibling and was in normal range. Amino acid profile in serum revealed decreased concentration of arginine, cytruline, homocysteine, PHE and ornithine. The patients suffer from severe symptoms of GERD such as vomiting, feeding problems instead of multistage therapy including Nissen fundoplication procedure as well as from epilepsy requires complex multidrug therapy. L-Arginine (200 mg/kg) and citrulline (100 mg/kg) were supplemented in the second sibling. The disease leads to premature apoptosis, so antioxidants (coenzyme Q, vitamin A and E) as well as carnitine were supplemented but without spectacular clinical results.
We provide clinical description of severe phenotype of de Barsy syndrome. Our molecular report broadens the spectrum of ALDH18A1 mutations causing de Barsy syndrome.
PDFShare this article
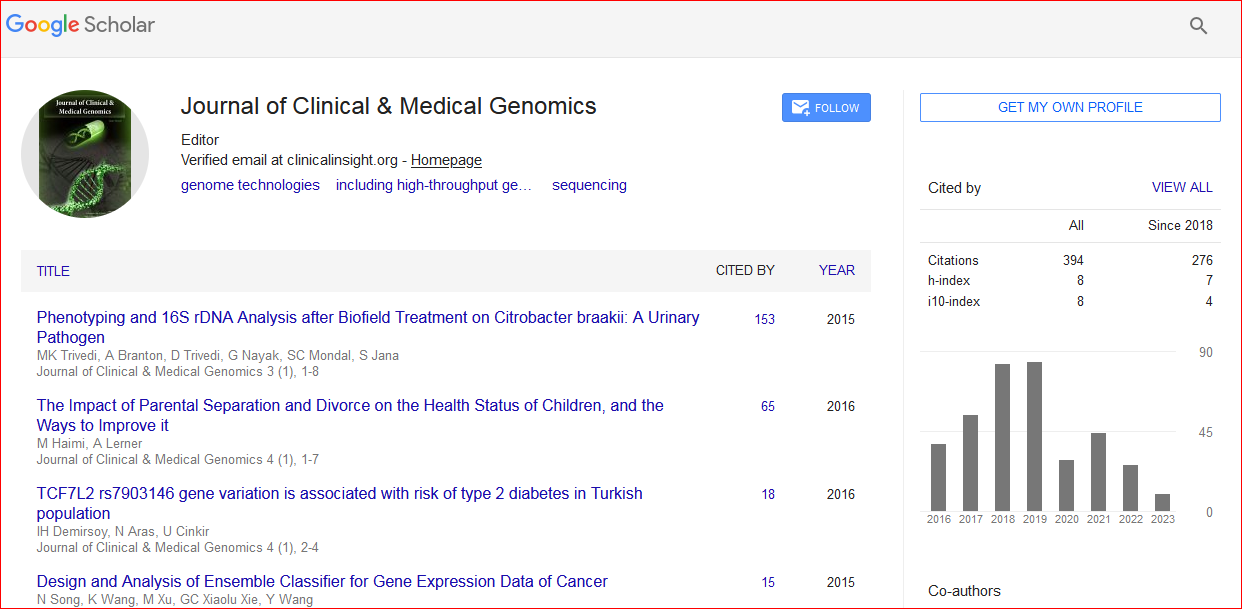
Journal of Clinical & Medical Genomics received 391 citations as per Google Scholar report