Kazue Takai
TAFRO syndrome is a systemic inflammatory disorder characterized by a constellation of symptoms; thrombocytopenia with reticulin fibrosis of bone marrow, anasarca including pleural effusion and ascites, fever, renal dysfunction, and organomegaly (hepatosplenomegaly and lymphadenopathy). Although several histopathological features of TAFRO syndrome resemble those of mixed type of multicentric Castleman disease (MCD), some cases of TAFRO syndrome don’t show any significant lymphadenopathy. In addition, several clinical and laboratory findings of TAFRO syndrome are different from those of MCD. The onset and clinical course of TAFRO syndrome may be acute or subacute, sometimes fetal, but its etiology, pathogenesis and specific marker is undetermined. Some patients have been successfully treated with corticosteroids, immunesuppressants including cyclosporine A, tocilizumab or rituximab, whereas others were refractory to treatment and succumbed to the disease. For contribution to the prompt diagnosis and appropriate treatment of TAFRO syndrome, the research team has defined its preliminary diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome. To promote the research on TAFRO syndrome, multicenter retrospective clinical study in Japan has been performed, and prospective study is being designed by a nation-wide research team on TAFRO syndrome.
PDFShare this article
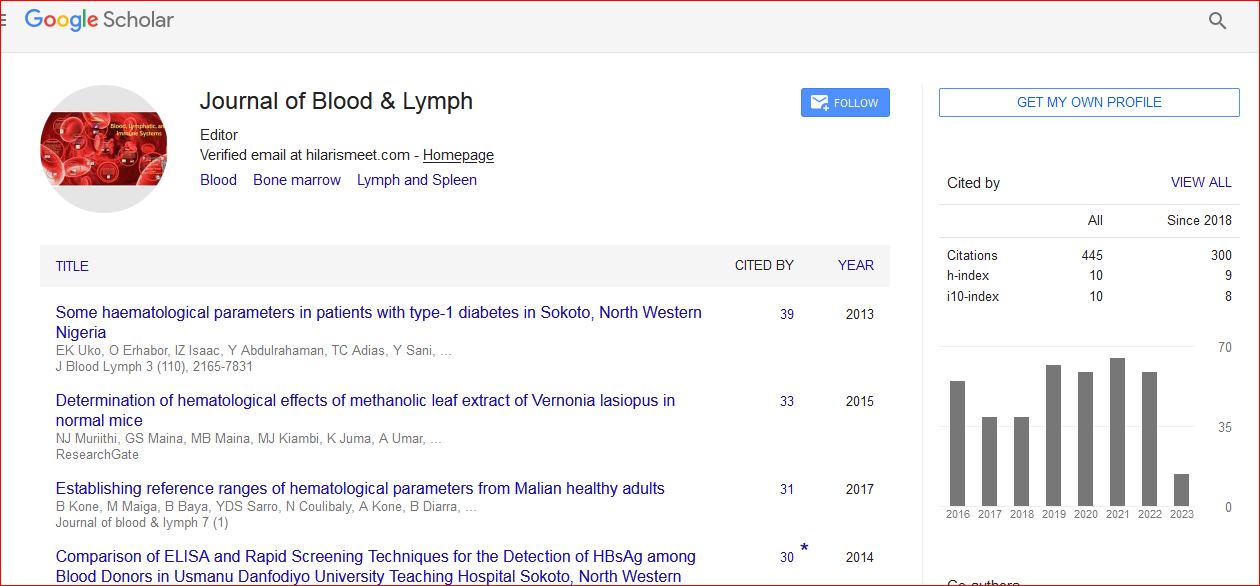
Journal of Blood & Lymph received 443 citations as per Google Scholar report