Ruofei Du, Donald Mercante, Lingling An and Zhide Fang
DOI: 10.4172/2155-6180.1000208
Background: Categorizing protein coding sequences into one family, if the proteins they encode perform the same biochemical function, and then tabulating the relative abundances among all the families, is a widely-adopted practice for functional profiling of a metagenomic sample. By homology searching of metagenomic sequencing reads against a protein database, the relative abundance of a family can be represented by the number of reads aligned to its members. However, it has been observed that, for short reads generated by next-generation sequencing platforms, some may be erroneously assigned to the functional families they are not associated to. This commonly occurred phenomenon is termed as cross-annotation. Current methods for functional profiling of a metagenomic sample use empirical cutoff values, to select the alignments and ignore such cross-annotation problem, or employ summarized equation to do a simple adjustment. Result: By introducing latent variables, we use the Probabilistic Latent Semantic Analysis to model the proportions of reads assigned to functional families in a metagenomic sample. The approach can be applied on a metagenomic sample after the list of the true functional families being obtained or estimated. It was implemented in metagenomic samples functionally characterized by the database of Clusters of Orthologous Groups of proteins, and successfully addressed the cross-annotation issue on both in vitro-simulated, bioinformatics tool simulated metagenomic samples, and a real-world data. Conclusions: Correcting cross-annotation will increase the accuracy of the functional profiling of a metagenome generated by short reads. It will further benefit differential abundance analysis of metagenomic samples under different conditions.
Heather M Bush, Jennifer Daddysman and Richard Charnigo
DOI: 10.4172/2155-6180.1000e130
Mahdi-Salim Saib, Julien Caudeville, Florence Carre, Olivier Ganry, Alain Trugeon and Andre Cicolella
DOI: 10.4172/2155-6180.1000200
Cancer is one of the leading causes of mortality. However, it is necessary to analyze this disease from different perspectives. Cancer mortality maps are used by public health officials to identify areas of excess and to guide surveillance and control activities. However, the interpretation of these maps is difficult due to the presence of extremely unreliable rates, which typically occur for sparsely populated areas and/or less frequent cancers. The analysis of the relationships between health data and risk factors is often hindered by the fact that these variables are frequently assessed at different geographical scales. Geostatistical techniques that have enabled the process of filtering noise from the maps of cancer mortality and estimating the risk at different scales were recently developed. This paper presents the application of Poisson kriging for the examination of the spatial distribution of cancer mortality in the "Picardy region, France". The aim of this study is to incorporate the size and shape of administrative units as well as the population density into the filtering of noisy mortality rates and to estimate the corresponding risk at a fine resolution.
Yu-Ting Chen, Diane Erwin and Dongfeng Wu
DOI: 10.4172/2155-6180.1000201
We applied a newly developed probability method to predict long term outcomes and over diagnosis in lung cancer screening using the Memorial Sloan-Kettering Cancer study (MSKC-LCSP) data. All participants were categorized into four mutually exclusive groups depending on their diagnosis status and ultimate disease status: symptom-freelife, no-early-detection, true-early-detection and over-diagnosis. Probability of each group is a function of the three key parameters: screening sensitivity, sojourn time in preclinical state and transition density from disease free to the preclinical state. We first obtained reliable and accurate estimates of these three key parameters using the MSKCLCSP data and likelihood function with a Bayesian approach, and then calculate the probability of each group by inserting these Bayesian posterior samples to the probability formulae, to predict future long term outcomes of lung cancer screening using chest x-ray. Human lifetime was treated as a random variable derived from US. Social Security Administration (SSA), so number of screening exams in the future is a random variable as well. The result shows that over diagnosis is not a big issue in lung cancer screening, given that it is only about 4.56% to 7.43% among the screendetected cases, depending on the age at the first screening.
Tabatabai MA, Li H, Eby WM, Kengwoung-Keumo JJ, Manne U, Bae S, Fouad M and Karan P Singh
DOI: 10.4172/2155-6180.1000202
In this paper we introduce new robust estimators for the logistic and probit regressions for binary, multinomial, nominal and ordinal data and apply these models to estimate the parameters when outliers or influential observations are present. Maximum likelihood estimates don’t behave well when outliers or influential observations are present. One remedy is to remove influential observations from the data and then apply the maximum likelihood technique on the deleted data. Another approach is to employ a robust technique that can handle outliers and influential observations without removing any observations from the data sets. The robustness of the method is tested using real and simulated data sets.
Miran A Jaffa, Mulugeta Gebregziabher and Ayad A Jaffa
DOI: 10.4172/2155-6180.1000203
Analysis of multivariate longitudinal data becomes complicated when the outcomes are of high dimension and informative right censoring is prevailing. Here, we propose a likelihood based approach for high dimensional outcomes wherein we jointly model the censoring process along with the slopes of the multivariate outcomes in the same likelihood function. We utilized pseudo likelihood function to generate parameter estimates for the population slopes and Empirical Bayes estimates for the individual slopes. The proposed approach was applied to jointly model longitudinal measures of blood urea nitrogen, plasma creatinine, and estimated glomerular filtration rate which are key markers of kidney function in a cohort of renal transplant patients followed from kidney transplant to kidney failure. Feasibility of the proposed joint model for high dimensional multivariate outcomes was successfully demonstrated and its performance was compared to that of a pairwise bivariate model. Our simulation study results suggested that there was a significant reduction in bias and mean squared errors associated with the joint model compared to the pairwise bivariate model.
Yingzhou Du, Chong Wang and Peng Liu
DOI: 10.4172/2155-6180.1000204
Pork and pork products have been identified as a significant source of Salmonella infection, which is a major public health concern. The contamination of Salmonella on pork can happen both on farms (before slaughter) and at abattoirs (after slaughter). Salmonella isolates were collected from both feces on farms and lymph nodes in the abattoir to determine if contamination at abattoirs can be linked back to the farms of origin. Molecular subtyping of the isolated Salmonella was performed using amplified fragment length polymorphism (AFLP), a Polymerase chain reaction-based, high-throughput, relatively inexpensive method. In this paper, we develop a permutation test for the genetic association of Salmonella isolated on-farm and at-abattoir using the AFLP data. Simulation studies show that the proposed permutation test controls the type I error rate appropriately as well as possesses high power. An application of the proposed permutation test to the real Salmonella ALFP data results in a p-value of 0.038 which shows strong evidence of association between Salmonella isolated on-farm and at-abattoir.
Shihong Zhu, Mai Zhou and Shaoceng Wei
DOI: 10.4172/2155-6180.1000206
Under the framework of the Cox model, it is often of interest to assess a subject’s survival prospect through the individualized predicted survival function, and the corresponding pointwise or simultaneous confidence bands as well. The standard approach to the confidence bands relies on the weak convergence of the estimated survival function to a Gaussian process. Such normal approximation based confidence band may have poor small sample coverage accuracy and generally requires an appropriate transformation to improve its performance. In this paper, we propose an empirical likelihood ratio based pointwise and simultaneous confidence bands that are transformation preserving and therefore eliminate the need of any transformations. The effectiveness of the proposed method is illustrated by a simulation study and an application to the Mayo Clinical primary biliary cirrhosis dataset.
Jing Wang, Sandeep Menon and Mark Chang
DOI: 10.4172/2155-6180.1000207
Adaptive clinical trial designs have been getting very popular in recent years. The PhRMA Working Group defines an adaptive design as a clinical study design that uses accumulating data to direct modification of aspects of the study as it continues, without undermining the validity and integrity of the [1]. These designs can assist in potentially accelerating clinical development and improving efficiency. However, the multiple interim looks and adaptive adjustments with the design can lead to inflation of type I error. Over the past decade, several statistical approaches have been proposed to control the inflation, some of which have been widely applied in practice. Some of these approaches include: error spending approach for classical group sequential plans [2-4]; Combination of p-values, such as Fisher’s combination test [5,6], Inverse Normal Method [7], sum of p-values approach [8]; conditional error function [9-11]; fixed weighting method [12]; variance spending method [13,14]; and multiple testing methodology such as closed test procedures [15-17].
Jianrong Wu and Xiaoping Xiong
DOI: 10.4172/2155-6180.1000209
In this paper, two parametric sequential tests are proposed for historical control trial designs under the Weibull model. The proposed tests are asymptotically normal with properties of Brownian motion. The sample size formulas and information times are derived for both tests. A multi-stage sequential procedure based on sequential conditional probability ratio test methodology is proposed for monitoring clinical trials against historical controls.
DOI: 10.4172/2155-6180.1000210
The one-sample log-rank test has been frequently used by epidemiologists to compare the survival of a sample to that of a demographically matched standard population. Recently, several researchers have shown that the one-sample log-rank test is conservative. In this article, a modified one-sample log-rank test is proposed and a sample size formula is derived based on its exact variance. Simulation results showed that the proposed test preserves the type I error well and is more efficient than the original one-sample log-rank test.
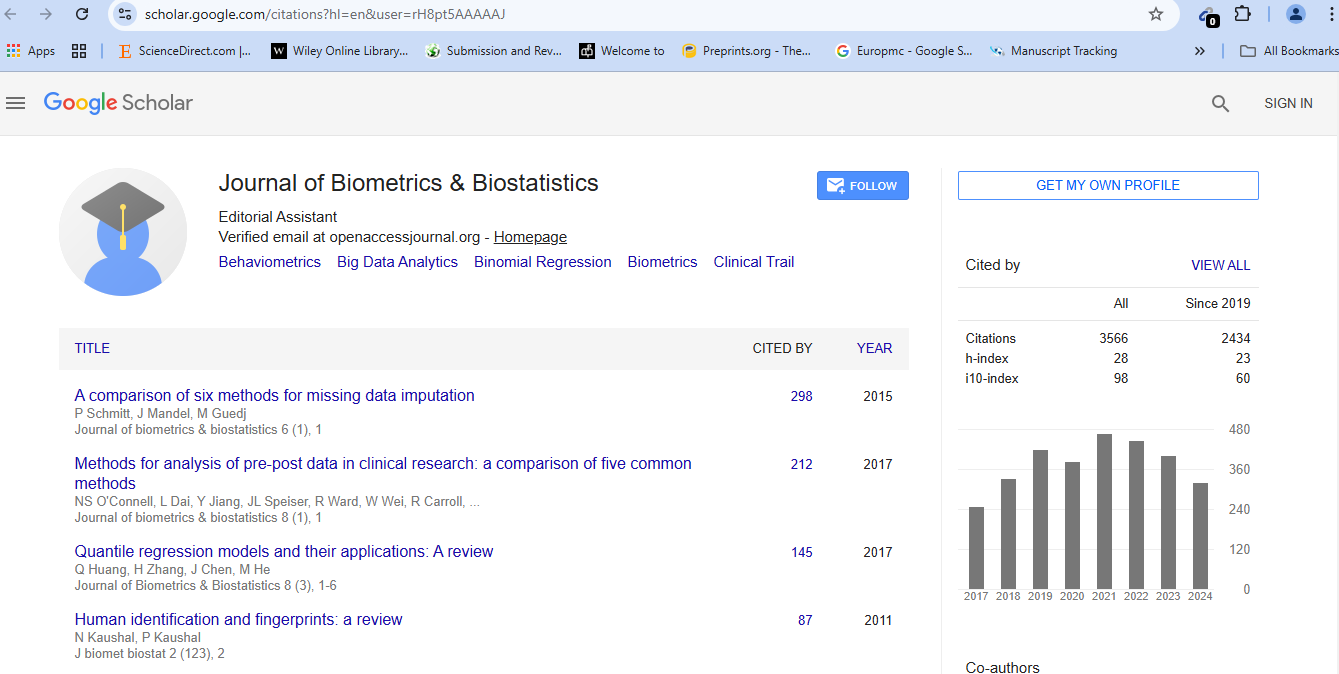
Journal of Biometrics & Biostatistics received 3496 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 