Fingerprint is a unique biometric feature of individual. It is also known that fingerprints have differences in male and female with respect to ridge line details. Some studies in machine learning investigate a relationship between fingerprint and gender. In these studies by analyzing the fingerprint we get important information such as age and gender of a person. Statistical studies have been made in different geographical areas to identify the relationship between fingerprint and gender. This paper illustrates gender classification based on fingerprints through various machine learning techniques like naïve Bayes method, Decision Tree and Support Vector Machine algorithms, KNN, PCA, Wilcoxon-Mann-Whitney Test, Friedman Test. This study introduces the concept of epidermal ridge, minutiae, ridge areas, ridge density etc., and compare above stated machine learning techniques, their limitations and strengths based on experimental results for gender classification based on fingerprints. This study can be useful for legislative cases and for researchers to devise new machine learning techniques with improved results.
Shin-Yi Tsai, Chon-Fu Lio, Leiyu Shi, Shou-Chuan Shih, Yi-Fang Chang, Yu-Tien Chen, Sheng-Kai Kevin Ma and Chien-Feng Kuo
Background: To explore the potential factors affecting time to discharge alive among burn patients and to determine the appropriateness of restrictive and liberal transfusion policies for burn patients. Study design and method: A retrospective analysis of 66 burn patients was conducted from 2013 to 2015. The average age was 26.7 and TBSA was 42.1% ( ± 25.9%). Data exploration of all dependent variables was performed to determine the normality and non-normally distributed variables were converted using Templeton’s two-step transformation involving percentile ranking. We assessed associations between significant clinical factors from and the outcome using Cox proportional hazards regression models with fixed and time-varying covariates. Impact of different transfusion threshold on the LOS was estimated by Cox proportional hazards regression and Kaplan-Meier curve. Results: A higher ABSI score (adj. HR, 0.28; P=0.017), present of bacteremia (adj. HR, 0.19; P=0.002) and pRBC transfusion (adj. HR, 0.55; P=0.001) were associated with significantly lower hazards of hospital discharge, suggesting a longer hospital stay. Further, the “restrictive” group also had a better outcome regarding the length of ICU stay (P=0.006) and hospital stay (P=0.003). There was a longer length of hospital stay in hemoglobin threshold greater than 8.5 g/ dl patients. (Log rank test, P=0.001). Transfusion threshold per se played an important role in extending the length of hospitalization (P=0.019). Conclusions: Restrictive RBC transfusion policy is more favorable to order appropriate blood components and helps the healthcare system to shorten LOS, reduce cost and complications.
Intervention effects on continuous longitudinal normal outcomes are often estimated in two-arm pre-post interventional studies with b≥1 pre- and k≥1 post-intervention measures using “Difference-in-Differences” (DD) analysis. Although randomization is preferred, non-randomized designs are often necessary due to practical constraints. Power/sample size estimation methods for non-randomized DD designs that incorporate the correlation structure of repeated measures are needed. We derive Generalized Least Squares (GLS) variance estimate of the intervention effect. For the commonly assumed compound symmetry (CS) correlation structure (where the correlation between all repeated measures is a constantρ) this leads to simple power and sample size estimation formulas that can be implemented using pencil and paper. Given a constrained number of total timepoints (T), having as close to possible equal number of pre-and postintervention timepoints (b=k) achieves greatest power. When planning a study with 7 or less timepoints, given large ρ(ρ≥0.6) in multiple baseline measures (b≥2) or ρ≥0.8 in a single baseline setting, the improvement in power from a randomized versus non-randomized DD design may be minor. Extensions to cluster study designs and incorporation of time invariant covariates are given. Applications to study planning are illustrated using three real examples with T=4 timepoints and ρ ranging from 0.55 to 0.75.
The introduction of Highly Active Anti Retro Viral Treatment has brought about a significant reduction in the morbidity and mortality of patients living with HIV/AIDS infection. However, the mortality rate of patients treated with Highly Active Anti Retro Viral Treatment is still high in developing country. The study has reviewed patient forms and follow-up cards of 1437 patients treated with Highly Active Anti Retro Viral Treatment in Dilchora Hospital in Dire Dawa from January, 2010 to December, 2016 G.C and used to identify factors leading to mortality and statistically modeling the survival of patients with HIV/AIDS treated under Highly Active Anti Retro Viral Treatment. Survival of patients was significantly related with gender, functional status, marital status, educational level, WHO clinical stage, place of residence and baseline CD4 cell count. Results of both Cox Proportional Hazard and parametric lognormal regression model revealed that; male, being bedridden, WHO clinical stage-IV, lived in rural residence and patients with lower baseline CD4 count had significantly higher risk of death or shorter survival time than their counterparts. Based on Akaike information criteria (AIC) parametric lognormal regression model best fits the dataset and used to predict survival experience of patients.
Qianfan Wu, Adel Boueiz, Alican Bozkurt, Arya Masoomi, Arya Masoomi, Allan Wang, Dawn L DeMeo, Scott T Weiss and Weiliang Qiu
Predicting disease status for a complex human disease using genomic data is an important, yet challenging, step in personalized medicine. Among many challenges, the so-called curse of dimensionality problem results in unsatisfied performances of many state-of-art machine learning algorithms. A major recent advance in machine learning is the rapid development of deep learning algorithms that can efficiently extract meaningful features from high-dimensional and complex datasets through a stacked and hierarchical learning process. Deep learning has shown breakthrough performance in several areas including image recognition, natural language processing, and speech recognition. However, the performance of deep learning in predicting disease status using genomic datasets is still not well studied. In this article, we performed a review on the four relevant articles that we found through our thorough literature search. All four articles first used auto-encoders to project high-dimensional genomic data to a low dimensional space and then applied the state-of-the-art machine learning algorithms to predict disease status based on the low-dimensional representations. These deep learning approaches outperformed existing prediction methods, such as prediction based on transcript-wise screening and prediction based on principal component analysis. The limitations of the current deep learning approach and possible improvements were also discussed.
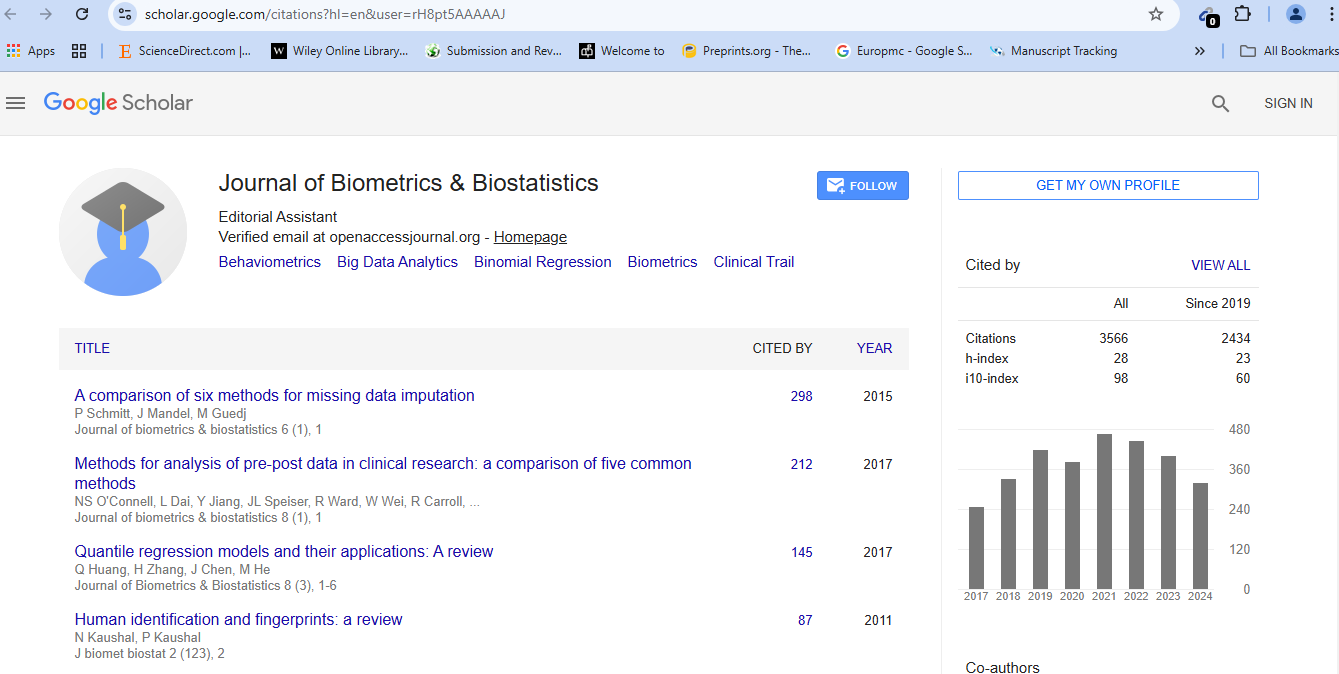
Journal of Biometrics & Biostatistics received 3496 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 