Research Article - (2021) Volume 9, Issue 6
Received: 25-Oct-2021
Published:
16-Nov-2021
Citation: Sanda, Srinath. “A Non-Coding Variant in SLC2A2 Predicts ?-Cell Loss in Type 1 Diabetes”. J Clin Med Genomics 9 (2021) 195.
Copyright: © 2021 Srinath Sanda. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Background: Type 1 diabetes patients lose residual β-cell function after diagnosis at variable rates affecting development of diabetes related complications. The cause of this variability is not understood. We hypothesized that common genetic variants in genes in the insulin secretion pathway would influence the natural history of type 1 diabetes. Methods: DNA samples and longitudinal insulin secretion data from 167 newly diagnosed type 1 diabetes patients were obtained. Participants were genotyped for common variants in insulin secretion pathway genes. Results: After accounting for age, high-risk HLA alleles, BMI Z-score, and gender in a mathematical model, a non-coding intronic variant (rs10513688) in the SLC2A2 gene correlated with rate of loss of insulin secretion over the first 12 months. To confirm the biological significance of this non-coding region we created a microdeletion in the homologous intronic region in mouse β-cells and observed reduced Slc2a2 and insulin expression suggesting a functional role for this non-coding region. Conclusion: This work suggests that a common genetic variant in SLC2A2 may associate with longitudinal decline in residual insulin secretion in patients with type 1 diabetes.
Type 1 diabetes• SLC2A2• SNP
Type 1 diabetes is an autoimmune disease characterized by pancreatic β-cell destruction [1]. Most patients have some endogenous insulin secretion remaining at the time of diagnosis, but the subsequent rate of loss is highly variable [2,3]. Patients with prolonged retention of endogenous insulin secretion after diagnosis have decreased retinopathy, nephropathy, and hypoglycemia compared to patients with more rapid loss of residual insulin secretion [4]. Therefore, understanding predictors of rapid loss of β-cell function is of clinical importance.
Variability in loss of endogenous insulin secretion is also relevant to current clinical trials that seek to modulate autoimmunity. Multiple intervention trials in new-onset type 1 diabetes participants (i.e. participants enrolled in clinical trials <100 days from diagnosis) aimed at preserving endogenous insulin secretion have been conducted over the past decade and have met with some success [5-10]. Interpreting these trials remains challenging since variables that predict the natural decline in insulin secretion in type 1 diabetes participants remain largely unknown and have not been incorporated into analysis of a candidate drug’s efficacy. Therefore, understanding the biology of endogenous β-cell loss after diagnosis could impact clinical research.
Age is the strongest predictor of β-cell failure with younger patients having greater rates of loss after diagnosis [2,3]. Analysis of genetic risk factors for type 1 diabetes such as high-risk human leukocyte antigen (HLA) alleles have shown conflicting results regarding their influence on longitudinal decline in insulin secretion, suggesting that while genes related to immune function identified by genome wide association studies may predispose individuals to develop disease, they may not influence longitudinal loss of β-cell function exclusively [11]. However, studies in individuals without diabetes suggest that variants in β-cell-specific genes may explain differences in glucose levels and insulin secretion in individuals without diabetes [12,13]. Therefore, we hypothesized that genes involved in the insulin secretion pathway in humans may predict the rate of decline in residual β-cell function in type 1 diabetes participants.
Study Population
DNA was obtained from participants (n=167), aged 7 to 46 years (mean age 16.9 years ± 8.76 years), who had participated in the placebo arms of one of five type 1 diabetes intervention studies conducted by Type 1 Diabetes TrialNet between 2006-2011 [6-10]. Participants in these studies were recruited from multiple sites in the USA and Canada. All studies were approved by local institutional review boards; all participants provided informed consent as outlined in the primary manuscripts for each study. Admission criteria were similar in all studies. All participants had at least one diabetes autoantibody associated with type 1 diabetes (insulin autoantibody if duration of insulin therapy was <7 days, glutamic acid decarboxylase 65, islet-cell antigen 512, or islet cell autoantibody), were enrolled within 100 days of type 1 diabetes diagnosis, and had a stimulated peak C-peptide level of at least 0.2 pmol/mL on a baseline mixed-meal tolerance test (MMTT). As part of the placebo arms, participants received no intervention but had intensive diabetes management provided by the study physicians and outside health care providers. Placebo participants underwent longitudinal 2- or 4-hour MMTTs as part of the study.
SNP selection
We chose to study common variants in genes related to insulin secretion. Using a curated pathway of 93 genes involved in regulating insulin secretion (R-HSA-422356) from the Reactome database (www.reactome.org), we identified 38 variants from these 93 genes that were covered by the Illumina Cardio-Metabo Chip and were: 1) intronic or within 2kb of the gene, and 2) did not violate Hardy-Weinberg equilibrium in the study population. We chose to restrict our SNP analysis to intronic or proximal variants in order to identify candidate genes that could plausibly contribute to loss of residual insulin secretion.
SNP genotyping
De-identified DNA samples were obtained from the TrialNet consortium for all participants and genotyped using the Illumina Cardio-Metabo Chip, a genotyping chip containing 195,945 single nucleotide polymorphisms (SNP) associated in genome-wide association studies with metabolic and cardiovascular endpoints, including glucose metabolism. Genotyping was performed using the manufacturer’s protocol. All participants were successfully typed for at least 90% of SNPs on the chip.
Metabolic data
Insulin secretion was quantified by calculating the area under the curve (AUC) for C-peptide collected during 2-hour MMTTs. For participants with a 4-hour MMTT, the AUC C-peptide was calculated based on the first two hours of data. Change in AUC C-peptide between months 0 and 6 and months 0 and 12 were calculated as (AUCmonth6 or 12 - AUCmonth0) / AUCmonth0.
HLA genotype
High risk HLA genotyping was performed by Type 1 Diabetes TrialNet for all study subjects and was classified based on the presence or absence of high risk DR3 (DRB*0301, DQA*0501, DQB*0201) or DR4 (DRB*0401, 0402, 0403, 0404, or 0405; DQA*0301 DQB*0302) alleles.
Cell culture
Min6 cells were donated by Dr. Gregory Ku at UC San Francisco and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 4.5 g/L glucose supplemented with 10% fetal bovine serum and 100x penicillin/ streptomycin. Cells were maintained at 37°C and 5% CO2. To subculture, the cells were treated with 1 mL 0.25% Trypsin/EDTA for 2 minutes at 37°C, then washed with 5 mL culture medium by centrifugation at 130x g for 5 minutes. All assays used Min6 cells grown to 70-80% confluence.
CRISPR Cas9 gene editing
Forward and reverse guide RNAs (5’-GTCTAAGAGGTGAGTCTAGG-3’ and 5’-AGCCTGTGTCACTCCAAATG-3’) were designed to delete an intronic region of the gene containing the target SNP. Equal parts tract RNA (IDT) and crRNA were hybridized at 37°C for 30 min to create guide RNAs at 50 μM, and then complexed with 20 μM Cas9 protein at 37°C for 15 minutes to create ribonucleoproteins (RNPs) at 10 μM. These RNPs were then added to 20,000 Min6 cells and electroporated using SF Cell Line 4D-Nucleofector X Kit with the 4D-Nucleofector 96-well Shuttle Device (Lonza) per manufacturer’s instructions. After expansion of each edited line, we confirmed genetic edits by PCR, agarose gel electrophoresis, and Sanger sequencing. Guide sequences shown in (Table 1).
| Oligo | Sequence (5'-3') |
|---|---|
| rs10513688 F primer | ATGCTTCACTCGGTCATGTC |
| rs10513688 R primer | GGAGGTTTGGGACCAGTAAG |
| CRISPR deletion gRNA 5 | GTCTAAGAGGTGAGTCTAGG |
| CRISPR deletion gRNA 6 | AGCCTGTGTCACTCCAAATG |
| mouse Ins2 exon 1 - intron 1 F primer | GGGGAGCGTGGCTTCTTCTA |
| mouse Ins2 exon 1 - intron 1 R primer | GGGGACAGAATTCAGTGGCA |
| mouse SIc2a2 F primer | ACACCGGAATGTTCTTAGCC |
| mouse SIc2a2 R primer | GTGAGAGAAGCCGAGGAAAG |
Table 1. Guide and primer RNA sequences used.
Real-time PCR
Total RNA was isolated from Min6 cells using PureLink RNA Mini Kit (Thermo Fisher) per manufacturer’s instructions and then reverse-transcribed into cDNA using SuperScript VILO cDNA Synthesis Kit (Thermo Fisher). qPCR experiments were performed using a CFX Connect Real-Time PCR Detection System (Bio-Rad). PCR products were detected using SYBR® Green Real- Time PCR Master Mixes (Thermo Fisher) with primer sequences provided in Table 1. PCR reactions were run in triplicate using an initial denaturation at 95°C for 30 seconds, followed by cycles of denaturation (95°C for 10 seconds) and primer annealing and transcript extension (60°C for 30 seconds) for 40 cycles. Relative expression of transcript was normalized to the average Ct value of beta-actin and GAPDH using the Livak method. Analysis was carried out using the CFX Manager Software (Bio-Rad). RNA sequences shown in Table 1.
Cell proliferation assay
Freshly harvested Min6 cells were resuspended in 5μM CFSE (Biolegend) in phosphate buffered saline (0.1%), then left to incubate at 37°C for 20 min. The cells were then washed, resuspended in culture medium, and seeded at a density of 50,000 cells/mL. Flow cytometric analysis was conducted on an LSRII (BD) flow cytometer at the UCSF Parnassus Flow Cytometry Core.
Statistical analysis
Quantile-quantile (Q-Q) plots across all QC’ed SNPs on the Cardio-Metabo Chip were used to evaluate presence of inflated test statistics. Multivariable linear regression was done to control for potential confounders in determining contribution of independent variables (gender, presence or absence of high risk DR3/DR4 alleles, BMI Z-score and insulin secretory genotype). Results were corrected for multiple comparisons using the method described by Benjamini and Hochberg [14]. An imputed q value of 0.2 was used for multiple comparisons analysis. For analysis of transcript data, triplicate results were analyzed using nonparametric tests (i.e. Mann-Whitney U tests) to compare fold changes between WT and edited cell lines. All analysis was done using R and GraphPad Prism.
Clinical characteristics
Samples, clinical data, and mixed meal tolerance data were obtained on placebo treated patients from 5 new-onset type 1 diabetes trials conducted by NIDDK TrialNet [6-10]. Out of 167 placebo treated participants, 10 were excluded because of missing MMTT data during the first year. Clinical characteristics of participants used in the regression analyses are shown in (Table 2).
| Study Participants (n=157) | |
|---|---|
| Gender | M=98 m(62%), F=59 (38%) |
| Age (y) | 16.97+/-8.97 |
| High- Risk DR3 alleles | Absent = 85 (54%) , Present=72 (46%) |
| High- Risk DR4 alleles | Abssent = 51 (32%), Present = 106 (68%) |
| Percent change in AUC C- peptide between month 0 and 6 | -21.91+/-48 |
| Percent change in AUC C- peptide between month 0 and 12 | -40.38+/-37.12 |
Table 2. Demographic, genetic and metabolic data of participants used in regression analysis.
Regression model analyses identify a noncoding variant in SLC2A2 that predicts decline in insulin secretion
After genotyping the study population using the Illumina Cardio- Metabochip, we first assessed whether th
ere was evidence of genomic inflation or population stratification. Principal component analysis of the data suggested absence of significant population stratification and we did not observe significant genomic inflation on the QQ plot (λ=1.039; Figure 1).

Figure 1. Quantile-quantile plot. (QQ-plot of raw p- values from multivariate analysis adjusting for population stratification (genomic inflation factor λ=1.039). Observed p-values are plotted on the y axis and expected p-values are plotted on the x axis.
We then selected variants from genes in the insulin secretory pathway as identified in the Reactome database (R-HSA-422356). Selected variants were either intronic or within 2kb of a gene in the insulin secretory pathway. A total of 38 variants from the Cardio-Metabochip met these criteria and were analyzed in multivariable linear regression model (Figure 2A). Given that age is a strong predictor of loss of insulin secretion, we constructed a model without age to maximize discovery of significant variants. Four variables (gender, BMI Z-score, DR3 status, and DR4 status) along with SNP genotypes were analyzed against percent decline of C-peptide over the first 6 months and the first 12 months after trial enrollment in a multivariable linear regression model. An intronic variant in the glucose transporter 2 gene, SLC2A2 (rs10513688 A/G), significantly correlated with rate of fall of insulin secretion over the first 12 months with a 19.1% decline in AUC C-peptide over the first year for every G allele (Figure 2B and Table 3, p value = 0.005). This relationship remained significant after multiple comparisons correction. No genotypes predicted rate of loss of insulin secretion at 6 months. Gender, body mass index (BMI) Z-score, DR3 status, and DR4 status were not significantly associated with rate of loss of insulin secretion at 6 months or 12 months after multiple comparisons adjustments.
| Variable | Estimate | p-value |
|---|---|---|
| Gender | -3.18 | 0.6 |
| BMI | 2.01 | 0.52 |
| DR3 | -10.17 | 0.08 |
| DR4 | 0.05 | 0.99 |
| rs10513688 | -19.11 | 0.005 |
Table 3. A multiple regression model (including gender, BMI Z-score, HLA DR3 status, HLA DR4 status and SNP) was run to determine the effects of variables on decline in C-peptide at 12 months. Estimated effect size and p-values shown.

Figure 2. rs10513688 correlates with loss of insulin secretion. List of variants included in linear regression model.
We then included the rs10513688 SNP along with age in a regression model to ensure the effect of the variant on insulin secretion was still observed when accounting for age. As predicted, age strongly associated with rate of loss of insulin secretion over the first 12 months (p value = 2.33×10-5). The rs10513688 variant remained significant even when accounting for age, with a 17.5% decline in AUC C-peptide for every G allele present (p value = 0.006) suggesting that this region, even when accounting for age, BMI, high risk HLA alleles, and gender could influence loss of residual insulin secretion in type 1 diabetes patients (Table 4).
| Variable | Estimate | p-value |
|---|---|---|
| Age | 1.34 | 2.338*10-5 |
| rs10513588 | -17.47 | 0.006 |
Table 4. Linear regression model including rs10513688 and age. Estimated effect size and p-values shown.
CRISPR/Cas9-mediated microdeletion of rs10513688 gene region in Min6 cells
A comparable cohort of participants with type 1 diabetes and longitudinal insulin secretion was not available to validate the candidate variant. Since the glucose transporter Glut2 is expressed on the surface of murine β-cells, we interrogated the functional significance of the intronic variant using genetically edited mouse cell lines.
The knockout of Glut2 in mice leads to impaired insulin secretion but functional interrogation of the intronic region containing rs10513688 has not been performed before [15]. We hypothesized that rs10513688 may reside in a cis-regulatory element and sought to determine if the intronic region surrounding rs10513688 regulated Slc2a2 expression through in vitro genetic manipulation of murine Min6 cells (Figure 3A). Rather than engineering the exact single nucleotide change in mouse cells, we chose to delete a small region of intron since the function of a specific nucleotide in a noncoding region may differ between species. We identified the homologous region in the murine genome corresponding to the human rs10513688 region using a clustal pairwise alignment (Figure 3B). We then deleted a small region containing the candidate variant using CRISPR/Cas9 directed editing. We electroporated CRISPR/Cas9 ribonuclear protein components along with two different guide RNAs directed at PAM sequences flanking the homologous SNP to specifically engineer a 44-nucleotide microdeletion in the intron of Slc2a2 while avoiding any alterations to the coding sequence (Figure 3C and Table 1). Edits were confirmed by PCR, gel electrophoresis and Sanger sequencing.

Figure 3. CRISPR/Cas9-mediated 44-base pair deletion in Slc2a2 intron 4 in the Min6 cell line. A. A graphic representation of human SLC2A2 gene with location of SNP rs10513688 indicated by arrow. B. Clustal pairwise alignment of human and murine SLC2A2 in the region surrounding SNP rs10513688. * indicates residue homology. C. Sequence trace alignment of Min6 wild-type (WT) and edited Min6 (Min6 d44) compared to reference sequence. Guide RNAs used for CRISPR/Cas9 deletion are shown.
Intronic microdeletion of Slc2a2 impairs insulin expression in Min6 cells
We first investigated Slc2a2 expression after introducing the microdeletion. Min6 cells with this 44-nucleotide microdeletion (Min6 d44) showed significantly impaired basal Slc2a2 expression compared to wild-type cells (Figure 4A). To determine whether deleting this genomic region played a role in β-cell function, we examined both basal insulin expression and cell proliferation in Min6 WT and Min6 d44 lines. Deletion of this region correlated with significantly reduced basal insulin transcription (Figure 4B). In addition, CFSE dilution assays showed reduced proliferation of Min6 d44 compared to Min6 wild-type (Figure 4C) suggesting a possible role for Slc2a2 in β-cell viability.

Figure 4.Microdeletion in Slc2a2 intron 4 containing rs10513688 results in reduced Slc2a2 and Ins2 expression and impairs proliferation of Min6 cells.
Our study suggests that genotype of the non-coding rs10513688 variant in SLC2A2 associates with a more rapid decline of endogenous insulin secretion after diagnosis of type 1 diabetes. This non-coding region affects β-cell function as evidenced by reduced Slc2a2 and insulin expression. These observations suggest that clinically observed heterogeneity in residual insulin secretion after diagnosis in type 1 diabetes patients may reflect inter-individual genetic differences in β-cells.
SLC2A2 encodes for glucose transporter 2 (GLUT2), a low affinity glucose transporter mainly expressed in the intestine, kidney, liver and pancreatic β-cell [16,17]. It is not the most prevalent glucose transporter molecule expressed on the membranes of pancreatic β-cells but its importance in regulating insulin secretion is well established [18]. Knockout mice show deficient insulin secretion [15]. In humans, mutations in SLC2A2 are observed in patients with Fanconi-Bickel syndrome, a disorder of carbohydrate transport characterized by hypoinsulinemia, hyperglycemia, hepatorenal glycogen accumulation, and nephropathy [17,19,20]. In addition, variants in SLC2A2, have been linked to type 2 diabetes and HOMA-B measurements in large GWAS studies [21,22]. Therefore, there is evidence to suggest that GLUT2 impacts insulin secretion in humans and lends rationale to our observations.
Our functional analysis of the candidate variant highlights the need for additional work. There are limitations to testing human variants in a murine system, namely that sequence homology while conserved is not identical between the two species. Therefore, single nucleotide substitutions may not be preserved from species to species, which led to our approach of introducing small microdeletions in the intron. Additional studies in human cell lines or freshly isolated islets will be crucial to understanding the biological significance of this variant.
We hypothesize the region containing rs10513688 may be a cis-regulatory element. However, one obvious explanation for reduced SLC2A2 expression with our intronic microdeletion is that the variant may lead to alternative splicing of the SLC2A2 transcript. At least 6 known splice variant mRNAs are transcribed from the gene resulting in different predicted proteins with variable function [23]. However, based on splice site prediction algorithms, the rs10513688 variant does not appear to result in altered splice products. Future studies will investigate whether genotype at rs10513688 variant affects transcription factor binding and RNA splicing. In addition, it would be useful to analyze whether other SNPs in the SLC2A2 region also correlate with β-cell function in type 1 diabetes.
There are several obvious shortcomings of this study. The number of variants tested and the sample size were limited. Given these shortcomings a focused search on genes related to insulin secretion was performed to maximize discovery. Furthermore, analysis of common variants within 2kb of a gene may have failed to capture other more distant loci more closely associated with longitudinal loss of insulin secretion. In addition, while this analysis did include age as a contributor to decline in residual insulin secretion, there were only 2 patients less than 6 years of age available for analysis. Therefore, the role of this variant in very young patients with type 1 diabetes is unknown.
Identifying genetic predictors of the natural history of type 1 diabetes has important clinical implications. Given the connection between early loss of residual insulin secretion and microvascular complications, it is possible that subsets of type 1 diabetes participants with genetic variants predisposing them to more aggressive loss of insulin secretion can be identified resulting in earlier screening for, and prevention of, nephropathy and retinopathy [4]. Additionally, genetic profiling could be used to interpret the results of interventional trials that use retention of residual insulin secretion as a study endpoint. This study identifies a potential non-coding variant in SLC2A2 that may stratify patients in such a manner. Additional studies in larger cohorts are needed.
Ethics approval and consent to participate
All clinical trials were conducted with ethics approval and all participants provided informed consent prior to enrollment in the respective clinical trials. Permission to access the samples was granted by NIDDK TrialNet. Consent to participate in the study was waived by the IRB at UC San Francisco since no identifying information was used.
Consent for Publication
Not applicable.
Competing Interests
The authors state that they have no competing interests.
Funding
This work was funded by grants from the Hellman Fellows Fund, NIDDK 1DP3DK111914-01, and an Innovation Award from the Coles Family. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
AH acquired data, analyzed data, and drafted the manuscript. PJ analyzed data and drafted the manuscript. NZ analyzed data and revised the manuscript. SS designed the experiments, acquired data, analyzed data, and drafted the manuscript. All authors have reviewed and approved the manuscript.
The authors wish to thank Peter Bense and Rushika Pandya for assistance with data management and programming and Alex Marson and Theo Roth for technical advice on CRISPR-Cas9 editing. We also acknowledge the support of the Type 1 Diabetes TrialNet Pathway to Prevention Study Group, which identified study participants and provided samples and follow-up data for this study. The Type 1 Diabetes TrialNet Pathway to Prevention Study Group is a clinical trials network funded by the National Institutes of Health (NIH) through the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of Allergy and Infectious Diseases, and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, through cooperative agreements U01 DK061010, U01 DK061034, U01 DK061042, U01 DK061058, U01 DK085465, U01 DK085453, U01 DK085461, U01 DK085466, U01 DK085499, U01 DK085504, U01 DK085509, U01 DK103180, U01 DK103153, U01 DK085476, U01 DK103266, U01 DK103282, U01 DK106984, U01 DK106994, U01 DK107013, U01 DK107014, UC4 DK106993, and the JDRF. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the JDRF.
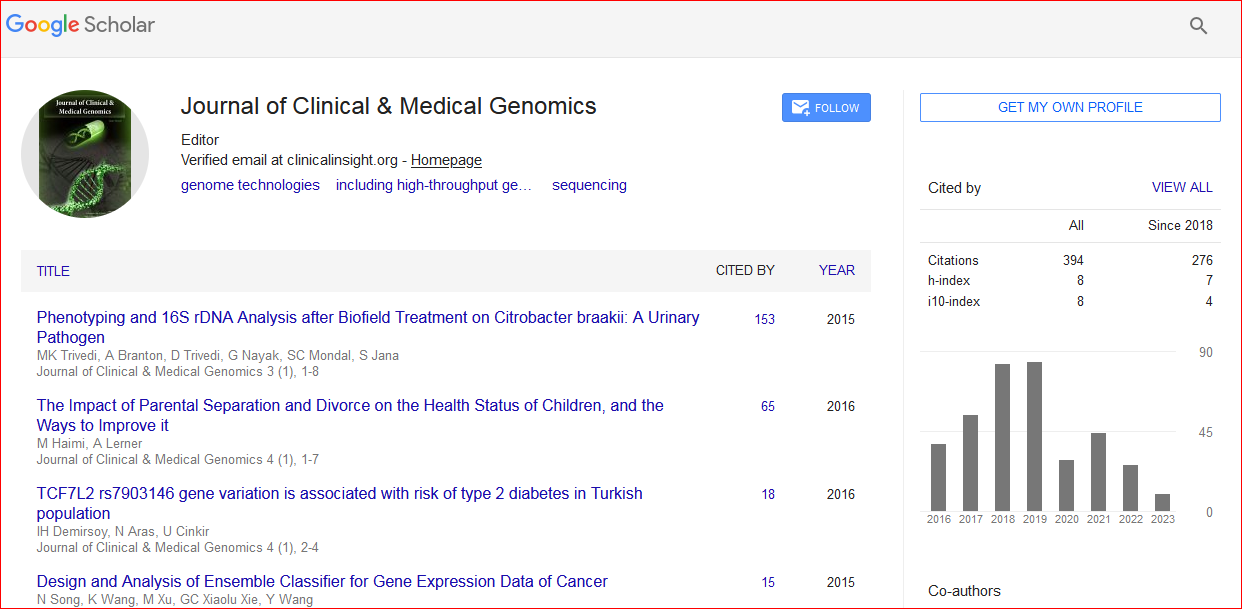
Journal of Clinical & Medical Genomics received 391 citations as per Google Scholar report