Review Article - (2020) Volume 4, Issue 5
Received: 05-May-2020
Published:
30-Oct-2020
, DOI: 10.37421/2684-4559.2020.4.131
Citation: Shreeya SR, Deepali VP, Arjun M and Vidya Niranjan. "A Pan Genome Review on Drug Resistance Mutations of Mycobacterium Tuberculosis; An Impending Threat to Healthcare". Clin Infect Dis 4 (2020) doi: 10.37421/jid.2020.4.131
Copyright: © 2020 Niranjan V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Tuberculosis, a bacterial infection caused by the organism Mycobacterium Tuberculosis, is a widespread disease infecting roughly one-fourth of the world’s population. Although a majority of these cases remain asymptomatic, tuberculosis continues to be the second- most common cause of death by infectious disease worldwide. A growing problem that is being exacerbated by the excessive use of multiple types of antibiotics is the emergence of drug resistant strains of tuberculosis. The number of Multiple Drug-Resistant Tuberculosis (MDR-TB) and Extensively Drug Resistant Tuberculosis (XDR-TB) strains are steadily increasing, rendering conventional treatment options ineffective. In order to circumvent the issue of drug-resistance, it is imperative that we understand both the mechanisms involved in resistance and the genetic mutations that cause it. In this review we delve into the important genes involved in drug resistance against common treatment options. We delineate the mechanism of resistance and specify the types and positions of mutations that have been observed in multiple studies worldwide. Finally, we discuss the relevance of the study to drug design, drug targeting and potentially forecasting future mutations by combining the mutation data with predictive analytics.
Mycobacterium Tuberculosis • Multi drug resistance • Whole genome sequencing • Mutation analysis • Single nucleotide polymorphisms
MTB: Mycobacterium Tuberculosis • MDRTB: Multi drug resistant tuberculosis • TB: Tuberculosis • LTBI: Latent tuberculosis bacterial infection • SNP: Single nucleotide polymorphism • INH: Isoniazid • MIC: Minimum inhibitory concentration • NADH: Nicotinamide adenine dinucelotide • RIF: Rifampicin • ORF: Open reading frame • RRDR: Rifampicin resistance determining region • XDRTB: Extensively drug resistant tuberculosis • NS: Non-Synonymous • EMB: Ethambutol • SM: Streptomycin • PZA: Pyrazinamide • POA: Pyrazinoic acid • PZAse: Pyrazinamidase • FQ: Fluoroquinolone • US FDA: United States Food and Drug Administration • RPT: Rifapentine • WGS: Whole genome sequencing
Tuberculosis is a highly infectious disease caused by Mycobacterium Tuberculosis, which spreads from the aerosols via spit, sneeze and cough of infected individual to healthy ones. More often, tuberculosis has no active and visible symptoms and is referred to as latent tuberculosis. This affects over a quarter of the world's population (as of 2018) and is considered as the most perilous form. Studies show that about 1% of the world’s population is affected by tuberculosis every year and in the year 2017, 10 million cases were recorded with about 1.6 million deaths. Tuberculosis is a dormant yet perilous disease which was once thought to be eradicated, due to the large-scale availability of TB vaccine, still largely plagues our society. This disease is more common in developing nations like India, Pakistan, China and the Philippines, with a record high of 95% of death occurring in these regions, owing to lack of awareness and unhygienic living conditions. Every year approximately 220,000 deaths are reported due to Tuberculosis, in India. Between 2006 and 2014, the disease cost Indian economy USD 340 billion. This public health problem is the world's largest epidemic. India bears a disproportionately large burden of the world's tuberculosis rates, with World Health organization (WHO) statistics for 2018 giving an estimated incidence figure of 2.2 million cases of TB for India out of a global incidence of 10 million cases. Historically, tuberculosis was referred to as consumption as it resulted is weight loss [1].
Tuberculosis infection is of two main types
Latent A. Tuberculosis Bacterial Infection (LTBI): Approximately 90% of the individuals who contract tuberculosis are asymptomatic. This means that they are not contagious and cannot spread it to others. The risk associated with LTBI, is that about 10% of these cases can progress to the active stage and if untreated can result in fatality. The cases only become active if the bacteria are reactivated. A person, who tests positive for LTBI, has to remain vigilant throughout their entire lifetime to ensure that they do not develop symptoms and become infectious, even if they have completed the course of medicine.
B. Active Tuberculosis: This kind of infection from mycobacterium is seen due to prolonged exposure to the bacteria by individuals with a weakened immune system. It is quite difficult to contract TB, a few minutes of contact is not sufficient to contract the same. Active tuberculosis affects about 10% of the individuals. They are highly contagious and can infect a healthy individual through aerosols spread through the air by coughing, sneezing or saliva. It is also referred to as Pulmonary TB [2].
Causative Agent-Mycobacterium Tuberculosis
M. tuberculosis is a pathogenic bacterium first discovered in 1882 by Robert Koch belongs to the Mycobacteriaceae family. It belongs to a complex that has at least 9 members: M. tuberculosis sensu stricto, M. africanum, M. canetti, M.Bovis, M.caprae, M. microti, M. pinnipedii, M. mungi and M. orygis [2]. It is a bacillus shaped bacterium, which is immobile, does not produce spores and aerobic in nature. It is small and takes about 15 h to 20 h to divide. It can survive weak or mild disinfectants and can live in the dry state for weeks. One key feature of the bacteria is its unique waxy surface coating of mycolic acid. The presence of Mycolic acid makes the bacteria resistant to Gram Staining, accounts for its ability to resist desiccation and most important is the reason behind its virulence. Since it cannot be detected by means of Gram Staining, acid fast stains like Ziehl Neelsen stains or Auramine stains (fluorescent stains) are commonly used [3]. It has been observed that the bacteria are rod shaped and are commonly seen sticking to each other due to presence of mycolic acid in their cell walls that make them sticky. This gives the appearance of a rope and is thus referred to as cording [4].
The most studied strain of MTB is H37 Rv. I was first isolated in 1905, New York from a 19-year-old patient, by Dr Edward R Baldwin. The genome of H37 Rv is composed of 4.4 × 106 bp and has around 4,000 genes. Annotation of the genome revealed that the bacterium has several unique features. More than 200 genes have been identified as encoding enzymes, playing a role in fatty acid metabolism. It has been predicted that approximately 100 of these genes have a role in the β-oxidation of fatty acids. The use fatty acids by such a large number of MTB enzymes may possibly be related to its ability to grow in infected host tissue, where the main source of carbon are fatty acids [5].
Drug resistance
Tuberculosis (TB) has regained its status as one of the major causes of death with approximately 3 million deaths annually, in the last decade. Further exacerbating the situation is the emergence of novel MTB strains which are resistant to few to all of the existing anti tubercular drugs. Major contributors to MDRTB outbreaks especially in public facilities and developing nations are the late recognition of drug resistance. Drug resistance in MTB is normally acquired by the bacteria either by mutation of the drug target or by drug titration due to target overproduction. MDRTB is primarily a result of mutation accumulation in the individual drug target genes. Currently TB is treated with a primary rigorous 2-month treatment composed of multiple antibiotics like Isoniazid (INH), Rifampicin (RIF), Pyrazinamide (PZA), and Streptomycin (SM) or Ethambutol (EMB). This is done to disallow the emergence of mutants resistant to any single drug. Therefore, mutant strains that are resistant to either of these drugs are major cause of concern. This situation leaves only drugs that mediocre to less than acceptable in their efficacy, have increased side effects, and have a higher mortality rate. The "MDR state" in mycobacteriology refers to simultaneous resistance to RIF and INH [6].
This paper will explore the various genetic and molecular basis for drug resistance of MTB towards the most utilized drugs for treatment, in order to identify potential ways to overcome the same.
Database used for analysis
Before conducting any research, it is important to familiarise oneself with the major open access resources that are available online, to obtain relevant and up to date information on tuberculosis. At present, there are three key tuberculosis resources. These are discussed below.
Tuberculosis database
Tuberculosis Database (TBDB) is a unified database that provides access to tuberculosis genomic data and resources which is pertinent to the discovery and advancement of tuberculosis biomarkers, vaccines and drugs. At present, the database contains the sequence data and annotations for 28 distinct Mycobacterium Tuberculosis strains and related bacteria. It houses pre- and post-publication gene-expression data of MTb and other related species. TBDB presently hosts data for approximately 1500 public tuberculosis microarrays and 260 Streptomyces arrays. Moreover, TBDB offers access to a compilation of software for comparative genomics and microarray analysis. It unifies M. tuberculosis genome annotation and geneexpression data with a compilation of analysis tools. It can be accessed here (http://www.tbdb.org/) [7].
a) Tuberculist Mycobrowser: The Mycobrowser (Mycobacterial browser), created and currently being annotated and maintained in the laboratory of Professor Stewart Cole, part of the Global Health Institute, is a complete genomic and proteomic data repository for pathogenic mycobacteria. It offers manually curated annotations and suitable tools to enable genomic and proteomic study of these organisms. The knowledge base of the mycobrowser unifies genome sequence details, drug and transcriptome data, information of proteins encoded for by the genes, operon and mutant annotation and structural and comparative genomics views in a meticulous fashion. This helps in the discovery and development of new diagnostic techniques, therapeutic and prophylactic measures which will help in combating the dangerous tuberculosis disease. Additionally, information on leprosy causing mycobacterium species is available here as well. It can be accessed here (https://mycobrowser.epfl.ch/) [8].
b) PATRIC: PATRIC is known as the Bacterial Bioinformatics Resource Centre. It is an information system intended to assist the work of the biomedical research community on bacterial infectious diseases through the unification of crucial pathogen information with rich data and analysis tools. PATRIC sharpens and enhances the scope of existing bacterial phylogenomic data from various sources, in order to save time and effort when conducting comparative analyses. It offers a large number of tools and services including data integration across several sources, type’s entities and organisms, a collection of annotated data obtained via RASTtk (an automated prokaryotic annotation system), High throughput computational services and a personal workspace [9].
c) RefSeq: The first step in any SNP analysis pipeline requires the alignment of a reference sequence with short sequence reads. The reference sequence is a reference genome obtained from the RefSeq repository which can be accessed through NCBI. The file format of the reference sequence should be FASTA [10,11].
d) SRA: Sequence reads are also necessary to execute an SNP analysis pipeline. Sequence reads can be obtained from the Sequence Read Archive (SRA) database which provides files which contain millions of short reads generated by high-throughput sequencing. The sizes of these reads tend to be less than a few hundred base pairs in length. Generally, the data from SRA can be downloaded in either the BAM format or FASTQ format [10,11].
Databases for Gene Expression Data: Gene expression data is used in various genome analysis techniques including SNP analysis, Genome Wide Association Studies (GWAS) and Differential Gene Expression Analysis (DGE). The GEO is a repository of gene expression data of various genome sequences obtained from different organisms. It is an international repository which typically contains microarray analysis data but also accepts data obtained from next generation sequencing and other high throughput techniques. There are many tools available on GEO NCBI, which can be used to identify differences in gene expression levels. Gene expression omnibus provides information on gene expression in the form of transcriptome data that is obtained from microarrays [3]. There are several analysis tools that are available on Gene Expression Omnibus that help obtain information regarding the important biological pathways the genes may be a part of. Most importantly, with the help of microarray data, we can obtain genes that are over expressed or under expressed as well as identify gene; gene interaction. Although GEO represents the most commonly used database to access gene expression data, various other gene expression databases, often specific to certain areas of research exist. For example, GENT2 is a database which explores gene expression patterns between normal and tumour tissues, and Gene Expression Database (GXD) provides gene expression data from the laboratory mouse [12].
Annotation
There are different tools used for annotation: SnpEff-A tool for annotation: SnpEff is a tool used for variant annotation and effect prediction. The inputs are predicted variants such as SNPs, MNPs, insertions and deletions. It is usually in Variant Call Format (VCF). The output is an annotated VCF file generated after SnpEff has analyzed the input variants. It also calculates the effects (e.g. amino acid change) that the variants produce on known genes. It recognizes many variants like SNP, insertions, deletions, multiple nucleotide polymorphisms etc. Annotation is done to know more about the variants than just the information present in a non-annotated VCF file. SnpEff provides both simple annotations (e.g. denoting the gene affected by each variant) and complex annotations (e.g. the effect of a non-coding variant on expression of a gene) [13].
a) ANNOVAR- A tool for annotation: ANNOVAR is a very effective software tool that has been designed to carry out the functional annotation of the genetic variants detected from diverse gnomes using quality upto- date information [14]. Diverse genomes include human genome hg18, hg19, hg38, as well as mouse, worm, fly etc. When provided with the list of variants along with information on the chromosome, start position, end position, reference nucleotide and observed nucleotides, ANNOVAR can carry out the following:
b) Gene-based annotation: Ascertain whether SNPs or CNVs result in protein coding changes and identification of the changes or impact on the amino acid sequences. Users can accommodatingly use RefSeq genes, ENSEMBL genes, UCSC genes, GENCODE genes, AceView genes etc. This is the most important use of Annovar for the use case described in this review paper.
c) Region-based annotation: Detects variants in precise genomic regions, for example, it can identify the conserved regions among several species, predicted transcription factor binding sites, segmental duplication regions, DNAse I hypersensitivity sites, RNA-Seq peaks and other annotations on genomic intervals.
d) Filter-based annotation: This is used in the identification of variants that are detailed in specific databases, such as in dbSNP, what the allele frequency in the 1000 Genome Project is and other annotations on specific mutations.
Screening for SNP
Genome Analysis Toolkit is a resource which provides access to several tools used to discover Single Nucleotide Polymorphisms and indels in data produced by Next Generation Sequencing (NGS). GATK is currently considered highly reliable and is the industry standard for variant calling. Many different modalities are provided to suit the type of sequences being analysed by the researcher. Some tools that help in screening for mutations especially SNPs are given as follows [15].
a) Haplotype Caller: Haplotype Caller is a tool that can call SNPs and indels at the same time. When the program approaches a region that shows signs of variation, it reassembles the reads in that region after discarding existing information about the mapping. Thus, HaplotypeCaller is more accurate to call regions which are difficult to call. This tool can handle pooled experiment data as well as non-diploid organisms.
Splice junctions, which make RNAseq analysis difficult for many variant callers, are well handled by HaplotypeCaller. The prerequisite for this is that the input read data must be pre- processed according to GATK recommendations. Before use in downstream analyses, VCFs should be filtered either by hard-filtering or variant recalibration.
The syntax of Haplotype Caller is given below: gatk Haplotype Caller -R filename.fa -I filename.bam -O filename.vcf
Where; I is the SAM/BAM/CRAM file containing the reads, -O is the VCF or GVCF file to which variants (raw, unfiltered SNP and indel calls) are to be written and R is the reference sequence file in fasta format. [15,16].
b) Select Variants: Select Variants is a tool that allows the selection of a subset of variants based on various criteria. This is done with respect to the intended analyses such as troubleshooting unexpected results, comparing and contrasting cases versus controls or, extracting non-variant or variant loci that meet desired requirements.
The various criteria by which a variant subset can be created from a complete callset include:
1. Inclusion thresholds on annotation values, e.g. "AF < 0.25" (sites with allele frequency less than 0.25). These criteria are referred to as "JEXL expressions".
2. Extracting samples based on either a complete pattern or sample name match.
3. Their type (e.g. INDELs), filtering status, evidence of mendelian violation and allelicity.
4. Give discordance or concordance tracks to exclude or include variants which are also present in other specified callsets.
The syntax of SelectVariants is given below: gatk SelectVariants -R filename.fa ;V filename.vcf -O filename.vcf
Where; O is output file to which selected subset of variants are to be written, -V is the VCF file containing all called variants and; R is the reference sequence in fasta format [15,16].
c) Variant Filtration: Variant Filtration is a tool which performs hard filtering on variant calls based on certain criteria. When the FILTER field has a value other than PASS, records are hard filtered. The filtered records are retained in the output by default. However, their removal can be requested in the command line.
The syntax for Variant Filtration is given below:
gatk Variant Filtration -R file name.fa -V filename.vcf --filter-name "filtername" -filter "QD<selectedvalue" -O filename.vcf
Where; R is the reference sequence in fasta format, -V is a VCF file containing variants, -O is the output file in which passing variants are annotated as PASS and failing variants are annotated with the names of the filters they failed. --filter-name accepts the name of the filter in quotations and filter specifies the threshold value for the variant to pass in quotations [15,16].
a) NGS role in SNP analysis: Next Generation Sequencing Data can be used to perform Single Nucleotide Polymorphism Analysis. This method of calling variants is widely used in the detection of drug resistant mutations in the genome of Mycobacterium Tuberculosis. The general workflow of an insilico SNP Analysis can be delineated as follows:
The first step to analyze SNPs is to either generate sequencing data or download the relevant data (SRA files and reference sequences) from online open source databases.
The next step involves the alignment of the short reads to the reference genome sequence. We must map the short-read sequences to a reference genome sequence to identify SNPs. One of the most popular tools used for DNA sequencing data is BWA (Burrows Wheeler Aligner) MEM tool which outputs the mapping as a Sequence Alignment/Map (SAM) file or a compressed version of that known as .bam file. Various other tools such as Bowtie, Bowtie2 and Novoalign can also be used.
The per-base quality scores estimated by base-calling methods are generally not well calibrated. Since these per-base quality scores play an important role in SNP detection and genotype calling, we must perform quality score recalibration analysis. This step can be implemented by tools such as ‘BaseRecalibrator’ and ‘ApplyBQSR’ available in GATK (Genome Analysis Toolkit).
Then we can call variants using a tool such as ‘Haplotype Caller’ which is a part of GATK. The output of this step is a raw Variant Calling File (VCF) which must be filtered before further use in downstream analysis. Various filtering tools such as ‘Select Variants’ and ‘Variant Filtration’ from GATK can be used for this. At the end of variant filtration we get a filtered VCF file containing the final list of variants. To easily comprehend the mutation data, it is important to annotate it. SnpEff is an example of an independent tool that is used for annotation and effect prediction [10,11].
Genome wide association study (GWAS)
A Genome-Wide Association Study (GWAS) is a method commonly used in genetics to correlate genetic variations with specific diseases. The technique involves examining the genomes obtained from numerous different people and then searching for genetic markers that can be used to predict the occurrence of a disease. Upon identification of the genetic markers, they are further used to comprehend the role of genes in disease occurrence ad thus advance prevention and treatment strategies.
In the context of Mycobacterium Tuberculosis, every year more strain are becoming drug resistant, with some being resistant to only first line drugs and some being resistant to more number of drugs. This is rendering the efforts to control Tuberculosis, useless. These polymorphisms can either be due to point mutations or Single Nucleotide Polymorphisms (SNPs) as seen in rpoB or they can be structural variants as seen in dfrAthyA double deletion. There are 7 established lineages of Mtb based on molecular typing.
These are prevalent in different parts of the world. GWAS, especially lineage-based studies can help in identification of novel drug targets. It is progressively being applied to pathogen research. It enables the identification of variants pam genome, associated with precise phenotypes. In order to prevent false associations, pathogen GWASs face the requirement to deal with the significantly elevated levels of population structure, seen in bacteria compared to humans, whilst enhancing sensitivity [17].
Whole genome sequencing
Whole-Genome Sequencing (WGS) has the ability to carry out highresolution genotyping and identification. It therefore encompasses more thorough information of pathogenic microorganisms and correctly identifies the Mycobacterium species. WGS can also simultaneously predict drug resistance built on known mechanisms of resistance at the genome level. In Recent Times, WGS has been demonstrated as a useful tool for Tb drug resistance prediction. Nevertheless, WGS has been used primarily in economically developed areas with low TB burden, so far. A wide collection of genetic mutations allow data from whole-genome sequencing to be used clinically for prediction of drug resistance, susceptibility, or for identification of drug phenotypes that so far cannot be predicted genetically. This methodology could be integrated into routine diagnostic workflows, replacing phenotypic drug-susceptibility testing while reporting drug resistance early [18].
Genetic and molecular analysis of drug resistance in MTB suggests that resistance is usually acquired by the bacilli either by alteration of the drug target through mutation (20) or by titration of the drug through overproduction of the target MDRTB results primarily from accumulation of mutations in individual drug target genes [6], In this section we discuss six common drug resistances, the mechanisms by which they occur and mutations that are associated with it.
Isoniazid resistance
Isoniazid or INH is a drug which is very active against MTB. It has an MIC in the range of 0.02 μg/ml to 0.06 μg/ml.
Mechanism of resistance: INH works by inhibiting the cell wall mycolic acids biosynthesis. This makes the bacteria susceptible to environmental factors and reactive oxygen radicals. An electron sink and catalaseperoxide (coded by katG) are responsible for the formation of an unstable electrophilic intermediate by the activation of INH. Since catalase-peroxide is the only enzyme which has the ability to activate INH any mutation in gene katG confers resistance by virtue of the product either being inactive or having a reduced activity as peroxide.
Mutations that occur in oxyR regulon, the regulon from which AhpC is transcribed divergently, are another possible explanation for isolates acquiring INH resistance. The inhA locus, is considered the main target for co-resistance to ethionamide and INH. This locus comprises 2 Open Reading Frames (ORFs) - inhA and orf1 which are separated by a noncoding region spanning 21-bp. In enterobacteria, inhA acts as a catalyst in a step in fatty acid synthesis and uses NADH as a cofactor. Iso-NAD, formed due to the action of katG on INH impedes the enzymatic activity of inhA, thus, blocking fatty acid synthesis. This could cause INH susceptibility. Some resistant strains show a T>G transversion, at position 280 of the inhA gene which causes the replacement of ser94 to ala94. This is considered to change the binding affinity between NAD(H) to inhA, resulting in INH resistance [6].
Drug resistant mutations: Complete deletion of the katG gene rarely occurs. Point mutations, insertions and deletions of up to 1 bases to 3 bases occur more commonly. A frequently observed mutation included G>T transversion in codon 463 where Leucine is substituted in place of Arginine and the MspI and NciI restriction site is lost [6]. Whole genome sequencing has been employed for identification of this mutation.
In a study conducted using samples from a hospital in Vietnam it was found that most, 71%, had the G944C (S315T) mutation. Apart from these, two isolates had G944T (S315I) mutations, two had G944A (S315N) mutations and one isolate had an A943G (S315G) mutation. KatG gene was deleted in one isolate. A 15C3T mutation in inhA was observed in ten isolates, a silent mutation in ahpC (C21A) was observed in one isolate and 11 isolates posessed a silent mutation in oxyR pseudogene (G37A) [19].
In another study on bacterial genetics 143 isoniazid-resistant strains were studied. 89.5% of them had at least one mutation in either the inhA promoter region or the katG gene and 11.2% had no resistance mutations in the sequenced regions. 64.3% of the strains had mutated katG gene and 40.6% strains possessed the S315T mutation. 32.2% strains showed inhA promoter mutations and a katG mutation was observed in 11(7.7%) of these cases. Mutations in the ahpC promoter are relatively uncommon, observed in 7.7% strains and these are compensatory, rather than directly resistance causing. [8,15]. In yet another study which used MTB samples from North India, katG(S315T) was seen in 90.5% of the isoniazid-resistant isolates, whereas 9.5% had no mutations in either katG or inhA [20].
Worldwide, 64% of all observed resistance to isoniazid was related to mutation of katG315. The mutation observed, second most frequently and which was seen in 19% of the resistant samples or isolates was that of inhA-15. These two mutations along with the ten of the most frequently occurring mutations in the inhA promoter and the ahpC-oxyR intergenic region account for 84% of worldwide phenotypic isoniazid resistance [21].
A study conducted in Shanghai, China, conclude that WGS is a very ppromising technique that can be utilized to predict the resistance of MTb towards isoniazid, rifampicin, ethambutol and other drugs. It was identified that the sensitivities towards isoniazid was 94.535 while for rifampicin and ethambutol it was 97% [22].
Rifampicin resistance
Rifampicin (RIF) is an anti-tuberculosis drug which has MICs in the range 0.1 μg to 0.2 μg.
Mechanism of resistance: rpoB codes for the ß subunit of RNA Polymerase. RIF specifically interacts with this subunit and hinders transcription. Mutations that occur in rpoB confer conformational changes causing improper binding of RIF and thus resistance [6].
Drug resistant mutations: The majority of mutations existed within an 81-bp core region and mainly constitutes changes in single nucleotide, resulting in single amino acid substitution. More than 70% of RIF-resistant isolates show changes in codons His526 and Ser531. Of the RIF-resistant mutations that do not fall in this 81-bp core region; other mechanisms are speculated. These include mutations in other subunits of RNA polymerase and changes to RIF permeability [6,23].
In the study conducted using samples from a hospital in Vietnam, the most common mutations were at codons 516 (15%), 531 (43%) and 526 (31%). Two insertions in codons 521 and 514 were identified and 10 of the isolates were found to have mutations at multiple codons. A mutation was observed in three isolates at codon 561 [20,24].
Mutations Observed by Country: India- Frequently mutated codons included rpoB 531, 526 and 516, where SNPs were prevalent.
Ser531Leu mutation, His526Tyr SNP and Asp516Val SNP are the most common SNPs noted. Double and triple mutations are also observed with Ser531Leu & His526Tyr SNPs and Ser531Leu, His526Tyr & Asp516Val SNPs respectively. Mutations also occur outside the 81-bp RRDR and include Asn413His, Asp 435Glu and Ala451Asp SNPs.
South Africa
In the Eastern Cape Province, resistance conferring mutations included SNPs Tyr42Asp, Leu92Ser, His87Gly, Leu457Pro, Val441Gly, Leu450Ser and Gly52Ala, most of them being outside RRDR of rpoB
In the KwaZulu-Natal Province, RIF resistance related mutations in MDR-TB isolates were Pro535Thr, His526Leu, Asp516Tyr, Ser531Leu, ILe572Met, Tyr645His, Leu533Pro. In case of XDR-TB isolates, the two main mutations associated with RIF resistance were L533P and D516G combination.
China
Non-synonymous (NS) mutations were observed at codon no. 509, 511, 516, 522, 526, 531, 533, 550 and 572. Two novel mutations were observed- Ser509Arg and Val550Leu. The bases of all the NS mutations are single base substitutions.
As per a study in Guizhou, rpoB Ser531Leu was the most common SNP, mutations at codon 526 were the second most common SNP and the third most common mutation was at codon 516 in rpoB gene.
Pakistan
The most common SNP observed was SNP S531L at rpoB gene and the second common mutation occurred at codon 516 with two SNPs Asp516Tyr and Asp516Val. Tyr528Tyr was the synonymous mutation found in this study and Leu533Pro and Ser512Ile were some other mutations. One isolate of MDR-TB carried double mutations at codon 516 and 512.
In a study with samples taken from Punjab, Pakistan, the most common rpoB NS mutations were Ser531Leu SNP and mutations at codon 516 [25,26].
Ethambutol resistance
Ethambutol (EMB) is a first-line anti-MTB drug. It is a synthetic compound [dextro-2,2'-(ethyldiimino)-di-1onol] with high levels of antimycobacterial effect. Mutation in Ethambutol genes were identified using the WGS method. It was then identified that WGS has a sensitivity to predict and discover mutation of 97% and specificity of 95.83%.
Mechanism of resistance: EMB administration causes rapid termination of mycolic acid transfer to the cell wall and equally fast accumulation of trehalose mono- and di-mycolates. In the cell wall, these mycolic acids get attached to 5'-hydroxyl groups of D-arabinose residues of arabinogalactan forming mycolyl-arabinogalactan-peptidoglycan complex. Disturbing arabinogalactan synthesis deters the creation of this complex and often leads to higher cell wall permeability. It has been shown that EMB specifically inhibits the transfer of arabinosyl. This suggests that the primary target for EMB is arabinosyl transferase. There are three ORFs in this locus- namely embA, embB, and embR. The embA and embB ORFs are separated from the embR ORF by a divergent promoter region of 178 bp. embR ORF region has been theorized to modulate embA and embB expression. The embB ORF does not have a ribosome binding site and is therefore translationally coupled with embA [6].
Drug resistant mutations: Studies among EMB-resistant isolates have indicated missense substitutions in the conserved embB codon 306 coding for Met. embB Met306 substitutions are the predominant substitutions. Strains of MTB with Met306Val and Met306Leu substitutions displayed a higher MIC in case of EMB, (40 μg/ml) compared to those for isolates with Met306Ile substitutions (20 μg/ml). Alterations in codon 306 may have a negative impact on the interaction of embB and EMB, resulting in a EMBresistance [6,23].
Another common substitution observed include substitution of Ser for Gly at codon 5 of embA. Additionally substitution of Asp for Gly at codon 406, Phe for Ser at codon 317 and His for Tyr at codon 334 of embB is observed [27].
Streptomycin resistance
Streptomycin was the first drug to be used to combat Mycobacterium Tuberculosis.
Mechanism of resistance: Streptomycin disrupts the bacterial activity by acting as a protein synthesis inhibitor. SM interferes with the binding of formyl-methionyl-tRNA to the 30S subunit, by binding to the small 16s rRNA of the 30S subunit of the bacterial ribosome. Inhibition of mRNA translation as well as misreading of codons disrupt cell functions, resulting in cell death [6,28].
The rpsL gene stabilizes bases of the 16S rRNA that are involved in translation, thereby playing an important role in translational accuracy. The SM drug affects the 16S rRNA, thereby inhibiting mRNA translation, ultimately leading to the death of the microorganism. Mutations in the rpsL gene can be attributed to the creation of a SM resistant strain of MTB. Single point mutations in S12 ribosomal proteins, coded by rpsL, along with mutations in rrs operon encoding the 16S rRNA confer the drug resistance. The single point mutations affect the 16S rRNA structure, such that SM cannot bind to it [29].
SM-resistance can be characterized by a pseudoknot formation in the 16S rRNA. Clinically isolated MTB strains showed base pairing between residue 524-526 and residue 504-507, which forms the pseudoknot.
The pseudoknot is further stabilized by G-U wobble base pairs between residues 522-501. From this, it has been concluded that the phenomena of Streptomycin resistance is a result of drug target alternation, not drug modification [14,30].
- Drug Resistant Mutations: In general, one of the lysine residues at positions 43 and 88 are replaced with arginine and threonine at the 43rd position, or arginine at the 88th position, in the rpsL gene. Additionally, an A>G transition is often observed at position 904 in the 16S rRNA, along with a single A>C transversion in the rpsL gene. This resulted in the substitution of Lysine-Glutamine in the 88th position. Since all of these mutations affect either the 16s rRNA or the ribosomal protein, mutation mediated drug resistance is conferred to MTB. It has been observed that mutations occurring in positions 491, 512, 516 within the 530 loop of rrs locus tend to be consistent with a Streptomycin-resistant phenotype [18,30].
As per a study conducted in China, the Mycobacterium Tuberculosis samples were first categorized as low-level Streptomycin-resistant and high-level Streptomycin resistant. The DNA was extracted, amplified and sequenced. Genotyping was performed by hybridization of biotin-labeled PCR-amplified DR locus against an array of 43 different immobilized DR spacers. The results of this study established that mutations in rpsL and rrs genes are associated with Streptomycin resistance in M. tuberculosis of 180 Streptomycin- resistant isolates, 83.3% of the strains harbored mutations in rrs or rpsL. This suggests a correlation between mutations in these genes and streptomycin-resistance. K88R (15.6%) and K43R (60%) were the two most abundant mutations in rpsL. Neither mutation was present in Streptomycin-susceptible strains, indicating that mutations K88R and K43R are involved in the development of Streptomycin resistance. It was also deduced that the nature of mutations in these loci is highly related to genotypes and geographical areas. Mutations in the gidB gene have been associated with low-level Streptomycin-resistance however, another study claimed to have detected mutations in gidB in susceptible M. tuberculosis isolates. In their study, low-level Streptomycin-resistance was observed to be highly related to deletion mutations. Therefore, mutation of the gidB gene has a potential role in Streptomycin-resistance [17].
A study in Iran also involved a similar procedure of drug susceptibility testing, DNA extraction and amplification. This was followed by spoligotyping. This study found that mutation rate in the rpsL gene was similar to that of the rrs gene (36.8%). Three types of mutations were identified in the rpsL gene of which substitutions at codon 88 (Lys→Met) and codon 43 (Lys→Arg) were the most common mutations found. The rate of mutation at codon 43 varies considerably between different geographical areas, accounting for 13.2% in Mexico, 25% in Brazil, 42.9% in North India, 70.4% in China and 80.4% of mutations in Singapore. Substitutions at codon 88 usually play a relatively minor role and occur less frequently than substitutions at codon 43. Point mutations at positions 516, 526, 865, and 907 were detected within the rrs gene. By sequencing it was found that Streptomycinsusceptible isolates were found to lack the abovementioned mutations in the rrs and rpsL genes [31].
Pyrazinamide resistance
PZA, which structurally resembles nicotinamide, is used as anti-MTB drug, mainly in short term chemotherapy. PZA is generally used as a sterilizer, as it does not possess great anti-bacterial properties. As a result, it is used in tandem with other drugs, like INH and RIF. The sterilizing power of PZA is very specific, it acts only on the MTB strain [6].
Mechanism of resistance: PZA along with INH is inactive as it enters the bacterial cell. The action of the PZAse enzyme converts PZA into pyrazinoic acid. POA is the active form of the drug, which acts as an sterilizing agent. The conversion of PZA into POA occurs in an acidic pH inside the bacterial body. While the cellular target for PZA has not been identified, it is theorized that enzymes involved in pyridine nucleotide biosynthesis are likely targets due to the similarity of PZA to nicotinamide. Further studies were conducted to verify the cause of drug resistance. The PZA resistant strains were transformed with the MTB pncA gene. This restored the previous susceptibility towards PZA in the microorganisms, indicating that mutations in pncA were responsible in creating a resistant strain [32].
Drug resistant mutations: Three main genes are involved in Pyrazinamide- resistance. The various mutations in these genes are described below:
pncA: The pncA genes found in resistant M. bovis variants were identified to have a single mutation at position 57. This single point mutation resulted in the substitution of Histidine to Aspartime. These single mutations were found in M. tuberculosis which showed resistance to PZA. Multiple mutations were observed in MTB: Cys138 replacement with Serine, Gln141 replacement with Proline, and Asp63 replacement with Histidine, along with the deletion of the G nucleotide at the 162 and 288 positions. These mutations led to the synthesis of an ineffective PZAse enzyme, which was incapable of converting the PZA into the active form. Mutations in pncA are diverse with respect to different geographical regions. 70; 97% of Pyrazinamide-resistant isolates of M. tuberculosis harbor mutations in either their pncA gene or a putative regulatory region [33,34].
RpsA: A recent study identified a novel target of Pyrazinoic Acid as the ribosomal protein S1 (RpsA). It was shown that binding of Pyrazinoic Acid to the 30S ribosomal protein S1 inhibits the trans-translation activity needed for the efficient synthesis of proteins. It was also found that the C-terminal region, where the DA438 deletion occurred in the PZA-resistant isolate, is the region that varies the most between Pyrazinamide-resistant and sensitive isolates. This indicates that changes in this region may alter Pyrazinamide susceptibility [33,34].
PanD: Some PZA-resistant strains lack mutations in rpsA and pncA genes as well as their flanking regions. Five low level PZA-resistant isolates without pncA or rpsA mutations were identified which instead had mutations in the panD gene. This gene encodes aspartate alpha-decarboxylase, a compound that is involved in synthesis of b-alanine. Additional sequencing analysis showed that the remaining PZA-resistant mutants in the study all harbored panD mutations which affected the C-terminus of the PanD protein. PanD M117I mutant was the most frequently observed mutation [34].
Fluoroquinolone resistance
Fluoroquinolones are a class of antibiotics which are synthetic derivatives of nalidixic, with a Minimum Inhibitory Concentrations (MICs) of 0.1 mcg/ml to 4 mcg/ml. FQs have been widely used in tandem with other drugs to combat TB.
Mechanism of resistance: FQs act on type II DNA topioisomerases called DNA gyrase. The DNA gyrase enzyme is coded by gyrA and gyrB genes. GyrA codes for introduction of negative supercoils in circular DNA. GyrB codes the coumarin-sensitive ATPase activity, as well as the heterotetramer structure of Gyr.
GyrA protein contains the cleavage activity, which is employed to determine quinolone sensitivity. GyrB contains the ATPase activity which is coumarin sensitive. Inhibition of DNA supercoiling and relaxation of Gyr is carried out by the Fluoroquinolone drug. FQs promote DNA cleavage with the help of Gyr, as quinolone drugs bind to single stranded DNA stronger than they do to double stranded DNA. As the re-ligation activity is disabled due to bond formation, the strands remain separated. This disrupts the transcriptional activity of the microorganism, leading to the death of the bacteria. Minimum Inhibitory Concentrations (MICs) of 0.1 mcg/ml to 4 mcg/ ml [35].
Drug resistant mutations: Expression studies of MTB identified mutations occurring in the gyr genes. The mutations, unlike in the case of different genes, were mostly clustered in a small area about 40 residues amino-terminal, near the active site tyrosine. Single point mutations were also observed for residues 88-94.
As bacterial strains become increasingly virulent and resistant to common drugs, it becomes important to modify/create a new medicine. Sparfloxacin is a new FQ derivative that possesses greater anti-MTB potency [35,36].
In a particular study, fluctuation analysis was performed and the mutational profiles of ofloxacin-resistant isolates in vitro were obtained. This paper concluded that Mtb genetic background has considerable role in the evolution of resistance to Fluroquinolones, that is, it can modulate the frequency of Fluroquinolone-Resistance acquisition. Fluroquinoloneresistant gyrA mutations that confer higher MICs, such as any gyrA gene mutation in codon D94 (with the exception of D94A) have been associated with poor treatment outcomes in Multi Drug Resistant-TB patients. The genetic background is therefore likely to contribute to variations in the treatment outcomes of patients when using Fluroquinolones as first-line drugs [35].
In another study, Mycobacterium Tuberculosis isolates were obtained from slant cultures of sputum, MIC value was determined for the isolates and whole genome sequencing was carried out. In this work, they characterized a set of Taiwanese TB samples to reveal novel insights into how Fluorquinolone-resistance emerges. Each patient carried both populations of resistant and sensitive organisms. Evidence of multiple gyrA alleles in 3 of these patients was observed. The study confirmed the simultaneous presence of several gyrA and gyrB alleles within a single patient, each conferring different levels of Ofloxacin-resistance. In every case, the resistant clones that were derived from the same patient carried a single SNP that conferred resistance [36].
Tuberculosis drug resistance is largely an anthropogenic problem that emerged due to alternation in the TB genome due to overuse of anti TB drugs. One of the primary and major reasons for this includes the noncompliance of the patients to the treatment process. The commonly followed practise to treat TB is a regimen of four drugs taken over sixmonths. This treatment regimen is extended up to 2 years with second line drugs to treat Multi Drug Resistant cases of TB. This results in challenges associated with treatment regimen compliance and can also lead to further mutations in the already MDR strains of TB. [37].
Identification of potent, novel and effective drugs and shorter treatment programs with high efficacy is the need of the hour. Clinal trials for various novel drugs and treatment regimens for tuberculosis are currently underway. Amongst these are old drugs (eg, daily high-dose RPT and higher dosages of RIF, linezolid and carbapenems) which are being repurposed and novel drugs (eg, bedaquiline, delamanid, and pretomanid [formerly PA-824]). Currently, High-dose, daily RPT is in phase 3 of clinical trials with focus on shortening the treatment period to around 3 months along with optimization studies (ClinicalTrials.gov identifier: NCT02410772). Testing for Pretomanid along with moxifloxacin and PZA as part of a combination regimen is underway, for the treatment of both drug-susceptible and drug-resistant tuberculosis (ClinicalTrials.gov identifier: NCT02193776). The US FDA approved Bedaquiline is also undergoing phase 2 clinical testing as part of a combination regimen along with pretomanid and PZA and/or clofazimine (ClinicalTrials.gov identifier: NCT01691534) [38].
Overall, it can be said that the tuberculosis therapeutics development field would largely profit with drug interaction studies, dosage studies for identification of novel drugs and improvement of the currently used drugs. As can be observed from section 2, most of the mutations are a result of deletions or additions of a single nucleotide, resulting in a frame shift. This is nothing but a single nucleotide polymorphism and they are quite common in MTB. While most are synonymous in nature, non-synonymous mutations play an important role in drug resistance like KatG, embB and embA or rpoB, results in conferring drug resistance upon the bacteria. A simple idea can be proposed to identify new drug targets; Carrying out an SNP analysis on MTB WGS collected from samples over 20 years or more years and from different geographical locations will help to identify the frequency at which SNP’s are occurring in the genes which play an important role in drug resistance or increasing virulence. With this we can identify new genes which may have been previously ignored due to lack of historical evidence. This can in turn help to identify new drug targets and or new mechanisms for drug action.
Authors thank Mr. Akshay Uttarkar for critical comments.
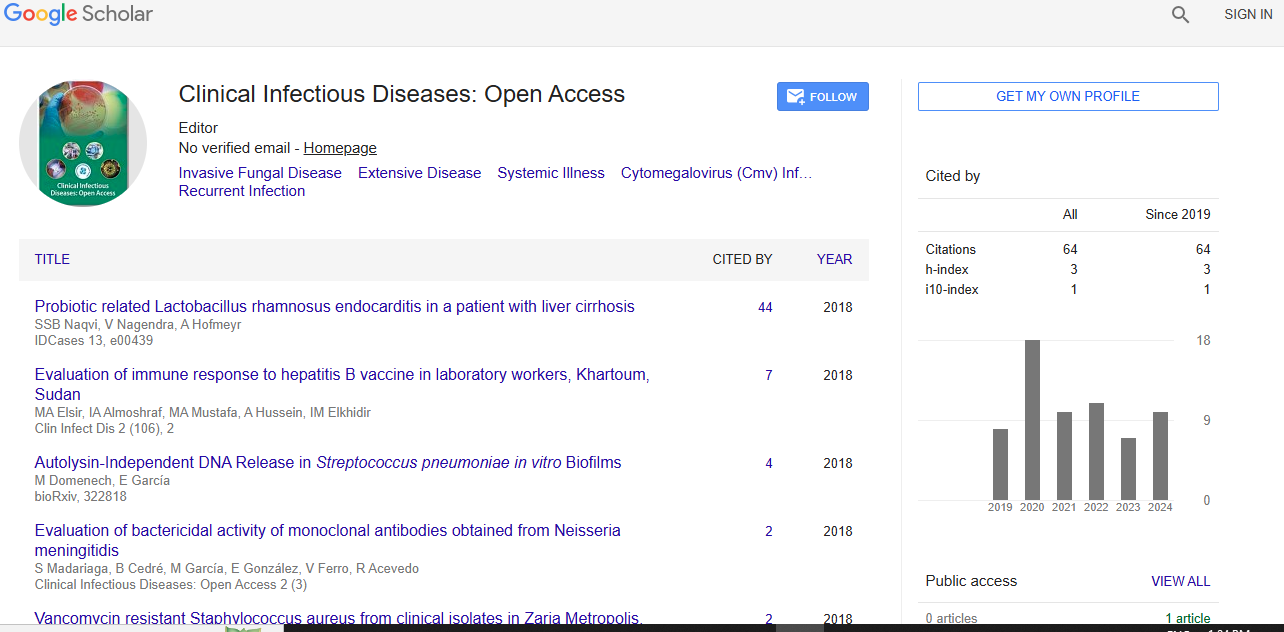
Clinical Infectious Diseases: Open Access received 1149 citations as per Google Scholar report