Research - (2020) Volume 8, Issue 8
Received: 01-Dec-2020
Published:
18-Dec-2020
Citation: Nirmala Koju, Abdoh Taleb, Zhou Jifang, Lv Ge, Ding Qilong. “α-Lipoic acid Prevents Angiotensin II-induced Endothelial Dysfunction via Antioxidant Effect and PI3K/Akt/eNOS Signaling Pathway” J Cardiovasc Dis Diagn 8 (2020) 8:417
Copyright: © 2020 Koju Nf, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Background: Oxidative stress is associated with endothelial dysfunction, the initial step in the pathogenesis of various cardiovascular disorders.
Methods: HUVECs were divided into 3 groups including control group, model group (cells treated with 10-6 M Ang II for 24 hr) and treatment groups (cells treated with 0.1 mM, 0.25 mM, 0.5 mM and 1 mM ALA for 30 minutes and further incubated with 10-6 M Ang II for 24 hrs). The cytoprotective effect of ALA against Ang II was tested by MTT assay, ROS generation was evaluated using Di-chloroflourescein (DCFH) and Mitosox Red, NO, ET-1 and antioxidant enzymes (SOD, GPx, CAT) and IDH) activity were analyzed by their respective kits. Furthermore, Western blot was used to detect the protein expression of Akt, p-Akt, eNOS, p-eNOS, Nrf2, PGC1-α, Sirt3 and Nox4. RT-PCR was used to detect the gene expression of Nox4 and eNOS.
Results: The results indicated that ALA in a dose-dependent manner lowered Ang II-induced loss in endothelial cell viability, reactive oxygen species and ET-1 production. Furthermore, ALA pre-treatment increased NO Level and antioxidant enzyme activity which is crucial for ROS elimination. The western blot results showed that Ang II markedly decreased Akt, eNOS, Nrf2, PGC1-α, Sirt3 protein expression which was enhanced with the pre-treatment of ALA that was further inhibited by a Wortmannin and L-NAME.
Conclusion: Our findings demonstrated that ALA possesses antioxidant activity against Ang II-induced oxidative stress partly by antagonizing AT1 receptor, suppressing Ang II-induced NADPH oxidase, increasing antioxidant enzyme activity and up-regulating PI3K/Akt/eNOS/NO dependent signaling pathway.
Alpha-lipoic acid • Angiotensin II • Oxidative stress • Endothelial dysfunction • Reactive oxygen species (ROS) • Antioxidant enzymes
Cardiovascular diseases (CVDs) represent a key cause of mortality in the world. Oxidative stress represents a difference between the formation of RONS (reactive oxygen and nitrogen species) and the ability of a biological system to detoxify these reactive intermediates or to repair the resulting oxidative damage [1]. Numerous studies evidenced that oxidative stress modifies various functional responses of vascular endothelial cells and stimulates inflammation, endothelial damage and cell death [2], accelerates the prevalence of cardiovascular diseases [3] such as hypertension, atherosclerosis, and myocardial infarction, as well as neurodegenerative disorders [4-7].
Several literatures have reported Mitochondria, xanthine oxidase, uncoupling of NOS and most especially the family of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases as the main sources of ROS [8,9]. Reactive oxygen species such as superoxide anion, hydrogen peroxide, hydroxyl radical, hypochlorous acid and peroxynitrite [10-16], can easily either take away electrons from other molecules or donate electrons to molecules [17]. Endothelial cells release a wide range of vaso-dilating and vasoconstricting substances such as nitric oxide, vascular adhesion molecule-1 (VCAM-1), E-selectin and intracellular adhesion molecule-1 (ICAM-1) [18,19]. Hence, endothelial dysfunction is the preliminary step in the pathogenesis of various cardiovascular disorders and related pathologies [10].
Angiotensin II (Ang II), an octa-peptide is formed after two subsequent reactions: first the enzymatic cleavage of angiotensinogen to angiotensin I by aspartyl protease renin. Then, angiotensin-converting enzyme (ACE) converts angiotensin I to angiotensin II [20]. When Ang II binds with its receptors (AT1 and AT2), different signalling pathways such as Akt/protein kinase B, p38 MAPK, ERK1/2, JAK and TYK are activated eventually enhancing the NADPH oxidase stimulation [21,22]. Angiotensin II has a pivotal role in ROS production, activation of apoptotic signalling pathways and thrombosis leading to endothelial dysfunction [23,24]. Akt kinase, also called a protein kinase B is a serine/ threonine kinase and its stimulation in PI3K/Akt signalling pathway depends on the upstream PI3K which leads to phosphorylation of Akt kinase. The phosphorylated Akt leads to activation of its downstream eNOS substrate and thus improved NO, which is an essential factor for maintaining the biological functions of endothelial cells [25,26]. To overcome the consequences of excess ROS, cells have evolved protective mechanisms such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidases (GPx), peroxidoxins and thioredoxin [27-30]. A balance between ROS generation and antioxidants is pivotal in maintaining redox homeostasis so that ROS can act as a messenger and regulate signalling pathways. Alpha lipoic acid (ALA) also known as thioctic acid is one of the potent antioxidants [31]. Literatures reported that ALA treatment diminished Ang II-induced NADPH oxidase activity, ROS production, and endothelial dysfunction; inhibited NF-kB mediated inflammatory responses; enhances the antioxidant enzymes such as superoxide dismutase, heme-oxygenase in rat skeletal muscle [32-34]. In this study, we hypothesized that ALA could attenuate the oxidative stress induced by Angiotensin II. Also, we examined whether the protective effect of ALA in endothelial cells that are exposed to oxidative stress is AT1R / Nox4 dependent and via-regulating PI3K/Akt/eNOS signalling pathway.
Ang II was purchased from Dalian Meilun Biotech Co., Ltd., ALA was purchased from Macklin Biochemical Co. Ltd. (Shanghai, China). eNOS, p-eNOS, Akt, p-Akt, Nrf2, PGC1-α, Sirt3 and β-actin antibodies were obtained from Wanlei-bio Co. Ltd. and Nox4 antibody was obtained from SAB biotic (Nanjing China). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody was purchased from Santa Cruz Biotechnology Inc (USA). Quantitative RT-PCR kit and SYBR Green Premix Ex Taq were obtained from Takara Biomedical Inc (Japan). Ang II powder and Alpha-lipoic acid were dissolved in dimethyl sulfoxide (DMSO) (less than 0.1%) and freshly diluted in culture media for all experiments.
Cell culture and treatment
HUVECs were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 U/ mL streptomycin. Cells were incubated at 37ºC with 95% humidity and 5% CO2. The medium was changed every 2-3 days until the cells reached 90% confluence and cells between 4 and 8 passages were used for the experiments. HUVECs were divided into control group, model group (incubated with 10-6 M Ang II alone for 24 hr) and treatment group (cells pre-treated with different concentrations of ALA (0.1 mM, 0.25 mM, 0.5 mM,1 mM) for 30 minutes and further incubation with Ang II (10-6 M) for 24 hr). In the western blot experiment, HUVECs were pre-incubated for 1h with AT1R, NADPH oxidase, eNOS, PI3K/ Akt inhibitors and Mitotempol.
Cytotoxicity assay in HUVECs
MTT test was performed to study the cytotoxicity effect of ALA in HUVECs. Briefly, cells were seeded in 96-well plates at a density of 5 × 103 cells/well for 24 hours. After 24 hours, cells were treated with different concentrations of ALA (0.1 mM, 0.25 mM, 0.5 mM and 1 mM) and one group was left untreated. The inhibitors Losartan, Apocynin, Wortmannin, L-NAME and Mitotempol were incubated for two hours. Then, 20 μL MTT solution was added to each group and then for 4 hours, cells were incubated at 37ºC. After 4 hour, DMEM containing MTT solution was discarded and 150 μL DMSO was added to each well. Finally, the absorbance was determined at 490 nm with a microplate spectrophotometer. The absorbance of untreated cells was regarded as 100% cell survival. Cell viability= (treated viable cells)/ (non-treated control viable cells) × 100%. Each experiment was performed in six replicates.
To determine the protective effect of ALA in the cell viability of HUVECs injured by Ang II, HUVECs were pre-treated with different concentrations of ALA for 30 minutes followed by 10-6 M Ang II for a further 24 hours. Losartan was incubated 1hr before the treatment of ALA and then the procedures were repeated as mentioned above.
Reactive Oxygen Species (ROS) Assay
HUVECs were incubated with different concentrations of ALA for 30 minutes and 10-6 M Ang II for 24 hours. In the groups using inhibitors, HUVECs were pre-incubated for 1 hr with AT1R, NADPH oxidase, eNOS, and PI3K/ Akt inhibitors and Mitotempol. After that, HUVECs were labelled with 10 μM DCFH-DA and incubated at 37ºC for 30 min. After three-time wash with PBS, the DCFH fluorescence was measured at excitation and emission wavelength 485 nm and 530 nm respectively, using an Olympus fluorescence microscope. Results were expressed as percentage of control (non-stimulated HUVECs) fluorescence intensity.
Measurement of mitochondrial ROS
For this experiment, HUVECs were cultured in six-well plates. Upon completion of the indicated treatment, cells were loaded with 5 μM Mitosox red and incubated for 10 min at 37ºC. Cells were washed gently three times with warm buffer. Fluorescence signals were recorded by a fluorescence microscope and images were visualized by a CLSM with an emission wavelength of 579 nm and the excitation wavelength of 510 nm, respectively.
Measurement of antioxidant activity
Antioxidants such as catalase (CAT), superoxide dismutase (SOD), glutathione peroxidase (GPx), and Isocitrate dehydrogenase (IDH2) were assessed according to the protocols provided with the assay kits. The enzyme activities were normalized to those of the control. NADPH oxidase inhibitor (Apocynin) was treated 1h before the treatment of ALA.
Nitric oxide (NO) assay
The supernatant fluid of HUVECs treated with ALA (0.1 mM, 0.25 mM, 0.5 mM, 1 mM) for 30 minutes and Ang II (10-6 M) for 24 hours was collected and then the concentrations of NO were measured with Griess reagent using total nitric oxide assay kit and the optical density was measured in 540 nm. To investigate the mechanism of NO production, inhibitors Losartan, Wortmannin and L-NAME were incubated 1hr before the treatment of alpha-lipoic acid. The amount of nitrite in the culture media was calculated using sodium nitrite as a reference standard following the instruction in the kit provided by the manufacturing company.
Determination of Endothelin 1 (ET-1) production
All the reagents were prepared as per the manufacturer’s instructions. The supernatant of the sample was collected and centrifuged at 2000-3000 rpm for 15 min. 50 μL from standard was taken and added to each well of the plate, then 50 μL from samples were added into each well, followed by 50 μL addition from biotinylated Ab to each well. Then the plate was sealed with a membrane and shaken gently, after that, it was incubated in the oven for 60 minutes. The washing process was done after very careful removal of the membrane. The washing solution was prepared and about 300 μL was added to each well repeatedly about 6 times according to the kit's instructions. After washing the next step was started by adding 50 μL of HRP and again sealing and washing. Furthermore, 50 μL from two solutions chromogenic A & B were added which was followed by the addition of 50 μL of stop solution. Finally, the absorbance was determined at 450 nm.
Quantitative Real-Time PCR
PCR was used to detect Nox4 and eNOS mRNA expression in each group. Total RNA was extracted from HUVECs by trizol reagent according to the manufacturer’s instructions (Invitrogen). cDNAs were synthesized with Prime Script RT Master Mix kit (Takara, Japan) following the manufacturer's instructions. cDNAs were used for PCR amplification using SYBR Premix Ex Taq TM (Takara, Japan) according to the manufacturer’s instructions on ABI Prism 7900 Sequence Detection System (Applied Biosystems, CA, and USA). β-actin was used as an internal control.
Primers sequences for PCR detection were as follows:
For Nox4: (Forward primer 5'-CAGATGTTGGGGCTAGGATTG -3', and
Reverse primer: 5'- GAGTGTTCGGCACATGGGTA-3').
For eNOS: (Forward primer 5'- TGATGGCGAAGCGAGTGAAG-3' and
Reverse primer: 5'- ACTCATCCATACACAGGACCC-3').
For β-actin: (Forward primer 5′-CGC AAA GAC CTG TAC GCC AAC-3′ and
Reverse primer: 5′-CAC GGA GTA CTT GCG CTC AGG-3’).
Western blot analysis
After treated by different experimental conditions with or without inhibitors, HUVECs were washed twice with ice-cold PBS and then lysed by using RIPA buffer containing 1% PMSF at 4ºC. The lysates were centrifuged at 12,000 g at 4ºC for 15 min and after that supernatants were collected, and the protein concentration of each sample was determined using the BCA Protein Assay kit according to the manufacturer's instructions. The lysates were denatured by boiling them in the SDS sample buffer. Then equal amounts of protein (50 g) from each group were loaded onto 10% SDS-PAGE and transferred to PVDF membranes. The membranes were blocked for 1 h in 5% non-fat dry milk and then incubated overnight at 4℃ with primary antibody against Nox4, Akt, p-Akt, eNOS, p-eNOS, Nrf2, PGC-1α, Sirt3 and β-actin as standard. After washing, the membranes were incubated with anti-IgG secondary antibody conjugated to horseradish peroxide for 1 hr at room temperature. Then the membranes were again washed in TBST for 30 min and exposed to enhanced chemiluminescence reagents. Densitometric analysis was performed to quantify the signal intensity. The relative density of each protein band was normalized to that of β-actin.
Image analysis was conducted with Image J software. Data were presented as the mean ± standard deviation (S.D.). One-way ANOVA followed by Dennett’s Multiple Comparison Test was used for the statistical significance of other groups compared with the control group and model group. A p-value of <0.05 was considered significant. All graphs were generated with Graph Pad Prism 7 software program.
An abnormality in ROS production and imbalance of endotheliumderived nitric oxide (NO) production has been long suggested to be the key pathogenic mechanism of the endothelial dysfunction [5,10,35] involving dysfunctional mitochondria that results in the development of clinical events, including vascular diseases and stroke [36]. Therefore, the effective protection of endothelial cells and reducing oxidative stress is beneficial in many experimental and clinical settings of CVD. In this study, we demonstrated that alpha-lipoic acid (ALA) protects HUVECs from Ang II-induced endothelial dysfunction.
Cytotoxic effect of alpha-lipoic acid in HUVECs
To investigate the antioxidant effect of alpha-lipoic acid, we first treated the HUVECS with four different concentrations of ALA (0.1 mM, 0.25 mM, 0.5 mM and 1 mM) with the motive to observe the cytotoxicity of ALA in endothelial cells. These tested concentrations of ALA were indicated as safe concentrations in several other in vitro studies where ALA has been implicated in inhibiting cytokine-mediated signalling, expression of adhesion molecule, and NO release in monocytes and endothelial cells [37-39]. With these experimental conditions, the result showed that the tested concentrations of ALA for 24 hours have not influenced the number of viable cells compared with the control group. Also, we have studied the effect of inhibitors such as Losartan, Apocynin, Wortmannin, L-NAME and Mitotempol in HUVECs by MTT assay. These inhibitors also did not show any significant cytotoxic activity in their respective concentrations (Figure 1).

Figure 1. Cell viability of HUVECs after incubation with the different concentrations of alpha-lipoic acid for 24 hrs. The inhibitors were treated with the relevant concentrations for 2 hrs. MTT assay was used to evaluate cell viability. Values are presented as means ± SD, n=3, NS=Non-Significant.
Effect of alpha-lipoic acid on Angiotensin II-induced HUVECs
Secondly, we studied the protective effect of alpha-lipoic acid against Angiotensin II-induced loss in the endothelial cell viability. As shown in Figure 2, the findings indicated that cell viability was diminished to approximately 66% when exposed to 10-6 M Angiotensin II for 24 hr, while it was increased up to 92% by pre-treatment of alpha-lipoic acid (1 mM). An in-vitro study on cytoprotective effect of ALA against hyperuricaemia induced endothelial dysfunction also revealed that 1 mM ALA significantly rescued the reduction in cell viability [40]. Similarly, another study also revealed that ALA significantly prevented cells against high glucose and bupivacaine toxicity respectively [41,42]. Thus, it is evidenced that ALA increased the survival rate of endothelial cells and provided the maximal protection against Ang II-induced injury. Besides, the pre-treatment of endothelial cells with angiotensin II receptor blocker (losartan) also markedly increased the cell viability. It clarifies that the AT1 receptor was activated by Ang II to induce oxidative stress and cell damage. Furthermore, the increase in cell viability by losartan and ALA (1 mM) were comparable. We believe that further preclinical research into the utility of losartan and ALA treatment, alone or in combination, may have significant potential in reducing Ang II-induced oxidative stress.

Figure 2. The concentration-dependent protective effect of alpha-lipoic acid in HUVECs after Ang II-induced cell injury. Losartan was incubated 1 hr before pre-treatment of ALA. MTT assay was accessed to evaluate the cell viability. Values are presented as mean ± SD, n=3. ###p˂0.001 vs. control group, *p˂0.05, **p˂0.01, ***p˂0.001 vs. model group.
Alpha-lipoic acid inhibited Ang II-induced ROS production
Various studies have evident that Ang II is responsible for ROS production leading to oxidative stress through different pathways. The incubation of endothelial cells with Ang II led to an increase in intracellular ROS production, which was significantly inhibited by pre-treatment with ALA in a concentrationdependent manner (Figure 3A). These findings more strongly suggest that a significant inhibition of Ang II-induced ROS generation by ALA may participate in restoring the viability of endothelial cells. However, to confirm mechanism how alpha-lipoic acid scavenge intracellular ROS generation induced by Ang II more clearly, we have used inhibitors like Apocynin (NADPH oxidase inhibitor), Losartan (AT1 receptor blocker), Wortmannin (PI3K inhibitor), L-NAME (eNOS inhibitor) and Mitotempol (mitochondria-targeted antioxidant). The ROS level was decreased in the group treated with Apocynin, Losartan and Mitotempol. We can hypothesize that the source of ROS in the present study can be Ang II-stimulated NADPH oxidase. Also, the ROS inhibition by Losartan, Apocynin and alpha-lipoic acid (1 mM) is comparable. This indicates that alpha-lipoic acid acts on oxidative stress in HUVECs by inhibition of NADPH oxidase. The inhibition might involve the blockade of the AT1 receptor. Similarly, mitochondria-targeted antioxidant Mitotempol and Alpha-lipoic acid (1 mM) has similar inhibition percentage (Figure 3C). Recent studies reported that the AT1R mediates many of its pathophysiological effects by stimulating ROS generation via a Nox-dependent mechanism [43]. Our results also advocate that both AT1 receptor activation and NADPH oxidase stimulation is involved in ROS generation. Besides, Mitotempol mediated decrease in intracellular ROS production recommends the ‘ROS induced ROS release’ (RIRR) mechanism. RIRR defines the mechanism in which ROS formed in one region activates ROS generation in another region of the cell. Generally, NADPH oxidase and mitochondria are considered as the major sources for ROS production. The decrease in intracellular ROS by a significant decrease in mitochondrial ROS by Mitotempol declares the ROS production by mitochondria and NADPH oxidase is bi-directional. Interestingly, the antioxidant effects of ALA against Ang II were abolished and a significant increase in ROS level was observed by Wortmannin and L-NAME treatment, indicating that ALA-regulated ROS elimination depends on PI3K/Akt activation (Figure 3B).

Figure 3A. Intercellular ROS generation in HUVECs was determined by measuring of DCFH Fluorescence. (A) Alpha-lipoic acid inhibits Ang II-induced ROS production in endothelial cells in a dose-dependent manner.

Figure 3B. The inhibitory mechanism was supposed to be via phosphatidylinositol 3-kinase PI3K/Akt/eNOS signaling pathway as Wortmannin and L-NAME reversed the protective action of ALA as indicated by representative images. Apocynin and Mitotempol also significantly reduced the ROS generation by Ang II.

Figure 3C. ROS fluorescent intensity index was presented as the percentage of the control group. Values presented as mean ± SD, n=3, ###p<0.001 vs. control group, **p<0.01 vs. model group, *** p<0.001 vs. model group, $$$p<0.001 vs. ALA (1mM) + model group.
Alpha-lipoic acid inhibited Mitochondrial ROS
Likewise, mitochondrial superoxide is also important for the development of hypertension, and antioxidant strategies specifically targeting mitochondria could have a therapeutic benefit [44,45]. ALA supplementation, however, can effectively increase mitochondrial targeting of superoxide scavenging and promote mitochondrial function in Ang II-induced endothelial dysfunction. The result showed that pre-incubation with different concentrations of ALA significantly reduced the Ang II-induced mitochondrial ROS. The mtROS production reduced from 383% in the cells treated only with Ang II (24 hr) to 133% in cells treated with 1 mM of ALA for 30 minutes, suggesting that ALA significantly improved the inhibition of mitochondrial ROS production in HUVECs lowering the chances of oxidative stress. Furthermore, we have treated the cells with inhibitors like Apocynin, Mitotempol and Losartan. Apocynin inhibited the mitochondrial ROS again indicating towards the ‘ROS induced ROS release mechanism’. It has been reported that Ang II via stimulation of AT1R, activates NADPH oxidase. This activation produces depolarized mitochondria, which further amplifies the mitochondrial generation of ROS [43]. In our study, Losartan also reduced the mitochondrial ROS justifying the above-mentioned fact that Ang II-induced AT1R stimulation was involved in the production of ROS in mitochondria. Also, the inhibition of mtROS by alpha-lipoic acid and Mitotempol was found to be analogous. These results demonstrated that ALA might inhibit ROS generation like Mitotempol (Figure 4). Similarly, our findings that superoxide production under Ang IIinduced cell injury was strongly inhibited by Apocynin and Mitotempol showed that both NADPH oxidase and mitochondria are likely sources of superoxide generation observed. Hence, ALA is a very beneficial antioxidant in lowering both intracellular and mitochondrial ROS and preventing cell damage from possible oxidative stress (Figures 4A and 4B).

Figure 4A. Mitochondrial ROS generation in HUVECs was determined by Mitosox red dye. ALA inhibits Ang II-induced mtROS production in endothelial cells in a dose-dependent manner. Also, inhibitors Apocynin, Losartan and Mitotempol lowered the mtROS significantly as compared to the model group as indicated by representative images.

Figure 4B. ROS fluorescence intensity index was presented as the percentage of the control group. Values presented as means ± SD, n=3, ###p<0.001 vs. control group, **p<0.01 vs. model group, ***p< 0.001 vs. model group.
Effect of ALA on antioxidant enzymes
The first line defence antioxidants which include superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) are crucial in maintaining the entire defence strategy of antioxidants and reducing the risk of oxidative stress-mediated diseases [46]. In oxidative stress, superoxide anion and hydrogen peroxide are formed and cannot be freely scavenged because of the low activities of catalase (CAT), glutathione peroxidase (GSH-Px) and (SOD) in the endothelial cell [47]. In our experiment, we studied activities of four different antioxidants SOD, CAT, GPx and IDH in HUVECs to determine the level of oxidative damage in HUVECs and to study the protective effect of Alpha-lipoic acid. As shown in Figures 5A, 5B, 5C and 5D), treatment with Ang II led to a significant decrease in the antioxidant enzyme level compared with the control group. In the current study, alpha-lipoic acid pre-treatment resulted in an increased enzymatic activity of IDH2, GSH-Px, CAT and SOD2 significantly in concentration-dependent manner (Figure 5). Our data demonstrated that Angiotensin II-induced endothelial dysfunction by decreasing the antioxidant enzyme activities, and preservation of these endogenous antioxidants may protect the HUVECs against oxidative stress [48]. It is necessary to identify whether enhanced ROS generation or diminished antioxidant activity accounts for an increased level of ROS. However, ROS generation was increased, and antioxidant enzyme activities were decreased in our study, we believe that Ang II-induced oxidative stress is attributed to increased ROS generation and diminished ROS scavenge. A study by Cao et al., evident the ability of ALA (low concentrations) in inducing antioxidant enzymes and resisting ROS induced cardiac cell injury [49]. Besides, we have used Apocynin to confirm the role of NADPH oxidase in the loss of antioxidant enzymes. In our study, we found that the Apocynin increased the antioxidant enzyme level as compared to the model group. This points toward the concept that the ROS production induced by NADPH oxidase might be the crucial factor for the diminished antioxidant activity. A study by Rizzetti et al. has also reported that Apocynin can not only prevent the reduction of SOD and GPx level but also increased their activity [50]. Within mitochondria, SOD catalyzes the conversion of H2O2 and O2. Thus, formed H2O2 is further decomposed into water by the act of catalase and GPx. During this process, GSH is oxidized to GSSG which is again reduced back to GSH by glutathione reductase. Therefore, well-organized action of various antioxidants is crucial for effective detoxification of ROS [49]. Similarly, the effect of ALA 1 mM and Apocynin is analogous indicating that ALA might have inhibited NADPH oxidase activation in the same manner as by Apocynin. Moreover, the detailed mechanism on how ALA increases antioxidant enzyme activities needs further investigation.

Figure 5. Concentration-dependent effects of alpha-lipoic acid on antioxidant enzymes (A) SOD (B) GPx (C) CAT (D) IDH in endothelial cells after Ang II-induced cell injury. Endothelial cells were pretreated with the indicated concentrations of ALA for 30 minutes and further incubated with Ang II for 24 hours. Apocynin was treated 1h before incubation of ALA. The antioxidant enzymes were studied as per the instructions of the manufacturing company. Values are represented as mean ± SD, n=3. ###p˂0.001 vs. control group, *˂0.05, **˂0.01, ***˂0.001 vs. model group.
Effect of alpha-lipoic acid on nitric oxide release
During this study, we assessed NO release and the results showed, as compared to the control group, the NO availability was significantly decreased in the Ang II-induced model groups. Following the experiment, pre-treatment with different concentrations of alpha-lipoic acid the Angiotensin II-induced effect in NO level was reversed (Figure 6). In the endothelium, ·NO produced by endothelial ·NO synthase (eNOS), regulates vasodilation and maintains basal vascular tone [50,51]. In the case of oxidative stress, the excess ROS not only decreases NO availability but also provokes apoptosis, inflammation by activating cell adhesion molecules and ultimately leads to cell damage [52-55]. Several studies have shown that eNOS is constitutively expressed in endothelium and is activated through PI3K/Akt-mediated phosphorylation, which leads to NO production [26,56,57]. Notably, several studies have demonstrated that LA increases eNOS activity [58-60], providing a potential mechanism that can explain its therapeutic effects on cardiovascular diseases. In our study, the model group Ang II (10-6 M) has reduced the NO level. ALA pre-treatment significantly increased the NO generation under Ang II-induced injury, but the effect was abolished by L-NAME and Wortmannin (Figure 6). Taken together with our present observations, our results demonstrated that ALA enhanced the function of eNOS impaired by oxidative stress by the PI3K pathway and increased NO availability. When HUVECs were treated with Losartan, the concentration of NO was increased significantly (the increment was comparable with the result of ALA 1 mM). In one in-vitro study, Marrero et al. [61] have demonstrated that the angiotensin II receptor may directly bind eNOS to inhibit its activity. Although the molecular pathway of angiotensin II action is not completely clear, our results lead to the hypothesis that ALA could interfere with the binding to angiotensin II receptor and prevent the angiotensin II-induced de-activation of eNOS [61].

Figure 6. Effect of alpha-lipoic acid on HUVECs in NO release in concentrationdependent manner. Wortmannin and L-NAME reversed the increment in NO by pretreatment of ALA. Losartan significantly increased NO availability. Values presented as mean ± SD, n=3, ###p<0.001 vs. control group, **p<0.01 vs. model group, ***<0.001 vs. model group $$p<0.01 vs. ALA + AngII group.
Effect of ALA on Endothelin-1 release
ET-1 is synthesized from a precursor, pre-proendothelin in several steps [62]. Various substances including Ang II and ROS can mediate ET-1 expression in endothelial and vascular smooth muscle cultured cells [63-66]. Our data revealed that the release of ET-1 from HUVECs when pre-treated with different concentrations of ALA was lowered in a dose-dependent manner as shown in Figure 7 whereas Ang II treatment alone for 24 hours significantly increased the ET-1 production. This increase in ET-1 release with Ang II treatment is comparable with the result of Moreau et al. In their experiment, the ET-1 level, blood pressure and vascular hypertrophy was increased with Ang II infusion [67]. We demonstrated that Apocynin and Mitotempol reduces the level of ET-1. These results showed the critical role of NADPH oxidase activity and mitochondrial ROS in the proliferation of ET-1. In one study by Chao et al. the expression level of p47phox and ET-1 release were significantly reduced in p47phox siRNA transfection and Apocynin treated cells as compared with the controls [68]. These results showed that ET-1 expression might depend on NADPH oxidase activation. In our study, Losartan markedly diminished the ET-1 production. This result is like another study where AT1 blocker losartan prevented Ang II-induced endothelial dysfunction and ET-1 production [69]. Ang II, a potent stimulator of ROS production, has been shown to increase ET-1 production in endothelial and vascular smooth muscle cells [70] as well as in vivo [69]. It was reported that ET-1 related vasoconstriction and hypertension were blocked with Ang II inhibition blocks [71]. Based on our findings, we can hypothesize that beneficial effects of ALA treatment to lower endothelial dysfunction may be mediated by decreased ET-1 levels in addition to an increase in NO production. ALA treatment has the potential to restore the balance between NO production and ET-1 secretion, because it both enhances the production of NO and inhibits secretion of ET-1.

Figure 7. Effect of alpha-lipoic acid on HUVECs ET-1 T release. Inhibitors like losartan, apocynin and Mitotempol also significantly reduced the ET-1 elevation. Values presented as mean ± SD, n=3 ###p<0.001 vs. control group, *p<0.05 vs. model group, **p<0.01 vs. model group, ***p<0.001 vs. model group.
Alpha-lipoic acid inhibited Nox4 over-expression via PI3K/Akt/eNOS dependent pathway
NADPH oxidase is the main source of ROS [72]. Inhibition of NADPH oxidase activity by its specific inhibitors VAS2870 and Apocynin, reverses endothelial dysfunction [73]. Numerous studies specify that Nox4, a dominant member of the NADPH oxidase family, is extremely expressed and active in endothelial cells [74,75], which associates in ROS production. We determined the underline molecular mechanisms accounting for the protective effects of ALA on HUVECs against Ang II, for that, we investigated the protein expression and gene expression of Nox4 mRNA. As shown in Figures 8A and 8B treatment with Ang II (10-6 M) for 24 hr significantly increased the level Nox4 protein expression compared to the control group (p<0.001). Alphalipoic acid pre-treatment in different concentrations (0.1 mM, 0.25 mM, 0.5 mM, 1 mM) for 30min inhibited Nox4 protein over-expression activated by Ang II in concentration-dependent manner (Figure 8B). Based on the above findings we can suggest that alpha-lipoic acid can decrease ROS generation and induce down-regulation of redox-sensitive signalling pathways in the vasculature tissue through inhibition of NADPH oxidase. In Figure 8A, we have observed the action of losartan (AT1R blocker) in Nox4 expression. The Nox4 was significantly downregulated by the treatment of losartan (p<0.001). This result evident that NADPH oxidase stimulation is regulated by AT1 receptor activation. Similarly, the inhibition expression of Nox4 by losartan and ALA (1 mM) are comparable indicating ALA might have chances of AT1 receptor blocking effects. Besides, when the cells were treated with losartan and ALA together, the result was more effective in lowering Nox4 expression than their effect. Therefore, it is worth studying whether they have a synergetic effect in the future (Figure 9).

Figure 8. (A) Effect of Losartan and ALA on Nox4 protein expression as determined by Western blotting and relative level of Nox4 protein expression after normalization to β-actin (B) Concentration-dependent effect of ALA on Nox4 expression in HUVECS after Ang II-induced injury as determined by Western blot analysis and relative level of Nox4 protein expression after normalization to β-actin. Values are presented as mean ± SD, n = 3. (###p<0.001 vs. control group, *p<0.05 vs. model group, **p<0.01 vs. model group, ***p<0.001 vs. model group).

Figure 9. ALA reduces Nox4 protein expression and protects Ang II-induced HUVECs via PI3K/Akt/eNOS pathway. Values are presented as means ± SD, n=3. (###p<0.001 vs. control group, ***p<0.001 vs. model group, $$$p<0.001 vs. ALA + Ang II group).
Likewise, the PI3K and eNOS inhibitor Wortmannin and L-NAME abolished the inhibitory activity of ALA against Ang II. The result of RT-PCR also showed that Nox4 mRNA expression was inhibited by the treatment of ALA. Losartan also inhibited the increase in Nox4 expression. The inhibition of ALA (1 mM) and Losartan was observed similarly supporting the result of western blot (Figure 10). Unfortunately, the Nox4 expression was again raised by Wortmannin and L-NAME (Figure 9). We have assumed this finding as ALA inhibits the Nox4 activity by PI3K/Akt/eNOS dependent pathway (Figure 9). However, the precise mechanism of ALA induced Nox4 down-regulation needs more investigation.

Figure 10. ALA reduces Nox4 RNA expression and protects Ang II-induced HUVECs via PI3K/Akt/eNOS pathway. Losartan also reduces Nox4 mRNA expression significantly as compared to the model group. Beta-actin was used as a reference gene. The results were interpreted using 2 -ΔΔCT method. Values are presented as means ± SD, n=3. (###p<0.001 vs. control group, ***p<0.001 vs. model group, $$$p<0.001 vs. ALA + Ang II group).
It is well known about role of AT1 receptor in vasoconstriction, cell proliferation [76,77], NF-kB stimulation [78], developing cardiac dysfunction [79,80] and hypertrophy [81]. Thus, it is obvious that Angiotensin receptor blockers (ARBs) would ameliorate the consequences of vascular oxidative stress. The AT1 antagonist, losartan, blocks most Ang II-mediated responses, and is clinically effective in the management of hypertension and arteriosclerosis [82,83]. Furthermore, our findings that Nox4 protein and mRNA expression was strongly inhibited by losartan also supports that the AT1 receptor was involved in stimulating NADPH oxidase (Figures 8A and 10). ALA (1 mM) produced a similar Nox4 inhibition as that caused by losartan. It has been reported that the enhanced expression of NADPH oxidase subunits is directly proportional to the increased production of ROS [84]. These results advocated that obstructing NADPH oxidase activation might be a pivotal mechanism in ALA-mediated protection against Ang II injury. However, the exact link between the PI3K/Akt signalling pathway and ALA inhibition of NADPH oxidase remains uncertain.
Effect of Alpha-lipoic acid on various protein expression induced by Angiotensin II
As shown in Figures 11C and 11D, treatment of Ang II (10-6 M) for 24 hours downregulated the protein expression of Akt and eNOS. When the groups were pre-treated with alpha-lipoic acid, the expression of Akt and eNOS proteins were increased significantly in concentration-dependent manner. Besides, we have studied the effect of alpha-lipoic acid on proteins Nrf2, PGC-1α and Sirt3 (Figures 11A, 11E and 11F). These proteins are pivotal in maintaining mitochondrial biogenesis, oxidative stress and antioxidant enzyme activity. The results showed that alpha-lipoic acid protected HUVECs from Ang II-induced damage and improved the expression of these proteins significantly in concentration-dependent manner.

Figure 11. Concentration-dependent effect of ALA on (A) Nrf2 (C) eNOS (D) Akt (E) Sirt3 (F) PGC1-α protein expression in endothelial cells after Ang II-induced injury (B) Relative bands of proteins as determined by Western blot analysis. (C-F) Optical densities were achieved after normalization to β- actin. Values are presented as mean ± SD, n=3. (###p<0.001 vs. control group, *p<0.05 vs. model group, **p<0.01 vs. model group, ***p<0.001 vs. model group).
Alpha-lipoic acid exerts protective effects on endothelial cells through Akt/eNOS pathways
In endothelial cells, phosphatidylinositol 3-kinase (PI3K) activates the downstream serine/threonine kinase Akt [85]. Akt stimulation induces the phosphorylation of the Akt, that is, the up-regulation of phospho-Akt. The phosphorylated Akt leads to activation of its downstream eNOS at Ser1177 substrate [86] which is mediated by kinase-dependent signalling pathways including PI3K/Akt, AMP-activated protein kinase (AMPK) and protein kinase
C (PKC), A (PKA), or G (PKA) [87-89]. It finally improves NO production, which is an essential factor to promote cell survival and maintain the biological functions of endothelial cells [87]. Therefore, the deregulation of Akt leads to major diseases such as cardiovascular, diabetes, cancer, and neurological diseases. In our study, the Akt and eNOS expression were downregulated by treatment with 10-6 M angiotensin II alone (Figures 11C and 11D). Compared with the model group, alpha-lipoic acid pre-treatment up-regulated the Akt and eNOS phosphorylation and increased the ratio p-Akt/Akt and p-eNOS/eNOS. On the other hand, LA-induced Akt and eNOS phosphorylation was completely abolished by PI3-kinase and eNOS inhibitors (Wortmannin and L-NAME respectively) (Figure 12) strongly suggesting that this phosphorylation is primarily mediated by the PI3-kinase/ Akt/eNOS signalling pathway.

Figure 12. The protective effect of ALA in HUVECs against Ang II-induced injury via PI3K/Akt/eNOS pathway (A) Western blot analysis for eNOS, p-eNOS, Akt and p-Akt. (B) Relative expression ratio of phosphorylated eNOS and Akt. Values are presented as mean ± SD, n = 3. (##p<0.01 vs. control group, ###p<0.001 vs. control group, **p<0.01 vs. model group ***p<0.001 vs. model group, $$p<0.01 vs. ALA + AngII group).
We have determined the eNOS mRNA expression when cells were treated with ALA, Losartan, Wortmannin and L-NAME. The PCR results showed that ALA and Losartan significantly increased the eNOS mRNA expression which was compromised in the Ang II-induced HUVECs injury model. But the Wortmannin and L-NAME abolished the increment in eNOS expression. This finding also focuses on the fact that eNOS activity is highly influenced by the AT1 receptor and is dependent on the PI3K/Akt pathway (Figure 13).

Figure 13. ALA increases eNOS mRNA expression and protects Ang II-induced HUVECs via PI3K/Akt/eNOS pathway. Losartan also increases eNOS mRNA expression significantly as compared to the model group. Beta-actin was used as a reference gene. The results were interpreted using the 2 -ΔΔCT method. Values are presented as means ± SD, n=3. (###p<0.001 vs. control group, **p<0.01 vs. model group ***p<0.001 vs. model group, $$p<0.01 vs. ALA+ Ang II group).
Likewise, we studied the role of PI3K/Akt/eNOS signalling in the protein expression of Nrf2, PGC1-α and Sirt3 by the western blot method. Interestingly, we found that all these three proteins were decreased significantly by Ang II and ALA pre-treatment has played a vital role in increasing their level as compared to the model group. But Wortmannin and L-NAME have downregulated Akt and eNOS phosphorylation and protein expression of Nrf2, PGC1-α and Sirt3 as compared to the ALA treatment group. This outlines the involvement of Akt/eNOS signalling pathway in ALA induced protective effects in HUVECs. Apocynin also increased the relative phosphorylation ratio of Akt and eNOS suggesting that NADPH activation by Ang II plays a crucial role in diminishing the phosphorylation of Akt and eNOS. (Figures 12A and 12B) and protein expression of Nrf2, PGC1-α and Sirt3 (Figures 14A, 14B, 14C and 14D). These also add the evidence that angiotensin was the vital factor for oxidative stress and alpha-lipoic acid protection against Ang II was mediated by inhibition of NADPH oxidase and AT1R inactivation.

Figure 14. The protective effect of ALA in HUVECs against Ang II-induced injury via PI3K/Akt/eNOS pathway (A) Protein bands of respective proteins determined by Western blot analysis. Relative expression of (B) Nrf2, (C) PGC1-α, (D) Sirt3.Values are presented as means ± SD, n=3. (###p<0.001 vs. control group, ***p<0.001 vs. model group, $$p<0.01 vs. ALA + AngII group, $$$p<0.001 vs. ALA + AngII group).
PGC-1 α, a key regulator of mitochondrial function [90], co-activates several nuclear transcription factors, which in turn regulate the expression of nuclear-encoded mitochondrial proteins. Sirt3, the primary mitochondrial deacetylase, plays a pivotal role in regulating ROS and multiple mitochondrial pathways [91]. So it is reasonable that the agents stimulating PGC-1α and Sirt3 expression in the endothelial cells are beneficial to eliminate ROS production and prevent the progress of cardiovascular complexities. A previous study also reported that PGC1α improved mouse Sirt3 activity in both hepatocytes and muscle cells, indicating that PGC1α acts as an endogenous regulator of Sirt3 [92]. A study by Song et al. reported that PGC-1α interacted with Nrf2 that bound to Sirt3 promoter to regulate Sirt3 expression and provided new insights into a critical Nrf2-PGC-1α- Sirt3 pathway in response to sodium fluorideinduced nephritic oxidative stress. In our study, we firstly found that the levels of PGC1α, Nrf2, and Sirt3 were all decreased in the endothelial cells treated with Ang II for 24 has compared to control group (Figures 11A, 11E & 11F). ALA treatment significantly reversed the Ang II induced decrease in PGC- 1α, Nrf2 and Sirt3 protein expression. These results confirm that LA affects mitochondrial metabolism and exerted antioxidant effects on endothelial cells damaged by oxidative stress partly by regulating the PGC1α - Nrf2- Sirt3 signalling pathway. Previous evidence showed that the PI3K/Akt pathway plays a crucial role in regulating Nrf2 activation and its subsequent nuclear translocation [93]. Moreover, the phosphorylation of Akt has been associated with the activation of Nrf2 [94]. In our study, the protein expression of Nrf2, PGC 1 α and Sirt3 by alpha-lipoic acid was disturbed by Wortmannin and L-NAME. It should be mentioned that increased phosphatidylinositol-3-kinase/Akt activity has been linked to the activation of the Nrf2-PGC 1α- Sirt3 pathway induced by alpha-lipoic acid as the inhibitors like Wortmannin and L-NAME disturbed the whole process (Figure 15). This result is comparable with another study which showed that melatonin protected liver cells against Na-F induced oxidative damage by triggering the PI3K/Akt-PGC1α signalling pathway and facilitating Sirt3 expression [95]. These results emphasize that the phosphatidylinositol 3-kinase (PI3K/Akt) pathway plays a major role in cell survival and is also required for Nrf2 and its downstream components activation [96].

Figure 15. Graphical abstract showing antioxidant effect of Alpha-lipoic acid in Angiotensin II-induced HUVECs via PI3K/Akt/eNOS pathway.
In conclusion, we can say, Ang II via AT1 receptor decreased cell viability, increased intracellular ROS, which were at least partly involved in Ang II-induced decreased phosphorylation of PI3K/Akt/eNOS pathway in HUVECs. Moreover, ALA is a potent antioxidant which in the present study, attenuated Ang II-induced oxidative stress and provided maximum protection of endothelial cells from oxidative stress through AT1R /Nox4/ROS and PI3K/ Akt signalling pathway. Thus, ALA may be considered as a future candidate for developing a novel therapeutic agent for preventing oxidative stress-induced CVDs. Our findings provide substantial evidence for new therapeutic options with AT1 receptor antagonists combined with antioxidants in the treatment of cardiovascular diseases associated with an increased activity of the reninangiotensin system. Nevertheless, additional investigations into the underlying molecular mechanisms of ALA in vivo are required to support our present findings.
Key Highlights
1) Oxidative stress involves excess ROS generation and reduced antioxidant capacity.
2) α-lipoic acid in a dose-dependently lowered Ang II-induced loss in cell viability.
3) ALA partly by antagonizing AT1 receptor down regulated ROS and ET-1 production.
4) α-lipoic acid up-regulates PI3K/Akt/eNOS/NO dependent signalling pathway
None.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
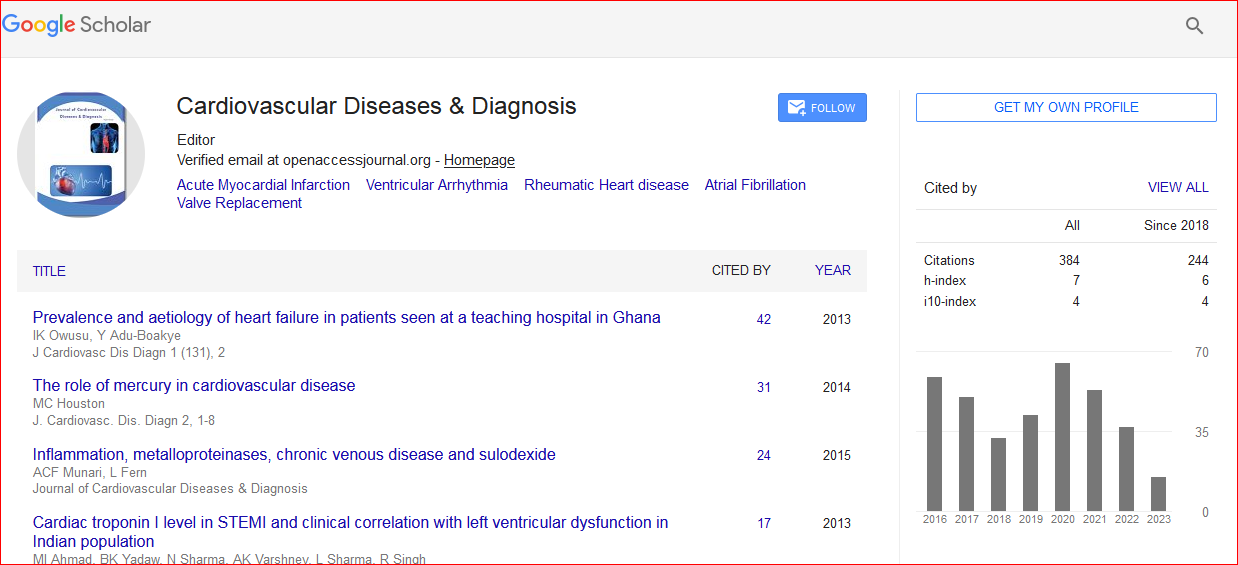
Cardiovascular Diseases & Diagnosis received 427 citations as per Google Scholar report