Editorial - (2022) Volume 13, Issue 1
Received: 05-Jan-2022, Manuscript No. jhmi-22-52248;
Editor assigned: 07-Jan-2022, Pre QC No. P-52248;
Reviewed: 10-Jan-2022, QC No. Q-52248;
Revised: 15-Jan-2022, Manuscript No. R-52248;
Published:
20-Jan-2022
, DOI: DOI: 10.37421/jhmi.2022.13. 401
Citation: Blue, Steven. "An Editorial Note on Cardiomyopathic Transformations in RBM20." J Health Med Informat 13 (2022): 401. DOI: 10.37421/jhmi.2022.13. 401
Copyright: © 2022 Blue S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
The most common reason for heart transplantation is dilated cardiomyopathy (DCM). Recent advances in DCM genetics have revealed over 50 DCM causal genes that are involved in a wide range of cellular processes and are high priority targets for precision therapies [1]. These studies have broadened the scope of cardiomyopathy research far beyond cardiac energy, conduction, and contractility. A growing body of evidence suggests that defects in RNA splicing and protein quality control play a role in the pathogenesis of heart failure. DCM can, in fact, be caused by mutations or dysregulation of multiple splicing factors [2]. Because the altered regulation of a single splicing factor can affect broad splicing regulatory networks, splicing factor mutations have a multifaceted effect, disrupting many cardiac signalling, transcriptional, and structural pathways.
RBM20 (RNA-binding motif protein 20) is a splicing regulator found primarily in the heart and skeletal muscle, where it plays an important role in cardiac physiology. RBM20 binds directly to the primary RNA (pre-mRNAs) of many cardiomyopathy-associated genes, where it ensures the proper production of adult protein isoforms via an exon exclusion process (i.e., splice isoforms associated with cardiac maturation). RBM20 autosomal dominant mutations account for up to 3% of DCM cases [3]. Furthermore, RBM20 DCM is highly penetrant, linked to life-threatening ventricular arrhythmias, and manifests at a younger age than DCM caused by mutations in other proteins (e.g., laminA/C or Titin). RBM20 DCM missense mutations are enriched in a five-amino acid stretch (RSRSP, which contains the R636S mutation) in the protein's arginine– serine-rich (RS) domain [4]. Surprisingly, nonsense or missense mutations in other parts of the 1227-amino acid-long protein are uncommon, implying that they are either undiagnosed due to a mild phenotype or are not tolerated.
WT Through binding to a UCUU consensus motif in adjacent introns, RBM20 has been shown to repress exon inclusion in key regulators of cardiac excitation–contraction coupling such as TTN, CAMK2D, and RYR2. Isolated cardiomyocytes (CMs) from RBM20-deficient rodents exhibit prolonged action potentials as well as striking calcium handling defects such as increased calcium transit amplitude, increased sarcoplasmic reticulum and diastolic calcium levels, and spontaneous calcium sparks. While loss of RBM20 expression in rodent models causes DCM, cardiac fibrosis, sudden death, and arrhythmias, few studies have focused on the precise role of mutant forms of the protein that correspond to human disease. Crucially, cross-linking immunoprecipitation (CLIP) sequencing approaches have only been used to examine RBM20 RNA-binding sites in wild-type (WT) rodent CMs and HEK293 cells.
Pathogenic RBM20 mutants have been studied in human, cell, and animal models. Analyses of resected human DCM hearts from people with RBM20 mutations (R636S or S635A) revealed intriguing global differences in spliceisoform and circular RNA expression, which are linked to cardiac contractility regulation. Calcium handling and splicing defects previously observed in rodent knockout (KO) models were discovered during CM differentiation of human-induced pluripotent stem cells (iPSCs) harbouring pathological forms of RBM20. Pigs were engineered with either heterozygous (HTZ) or homozygous (HMZ) R636S mutations in RBM20 in a recent impressive effort [5]. This model resulted in three critical findings:
(1) RBM20 R636S HMZ mutation causes highly penetrant neonatal lethality due to heart failure.
(2) Mutant RBM20 co-localizes with stress granules in the CM cytoplasm after sodium arsenate-induced metabolic stress and
(3) RBM20 undergoes an apparent liquid–liquid phase separation.
None.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
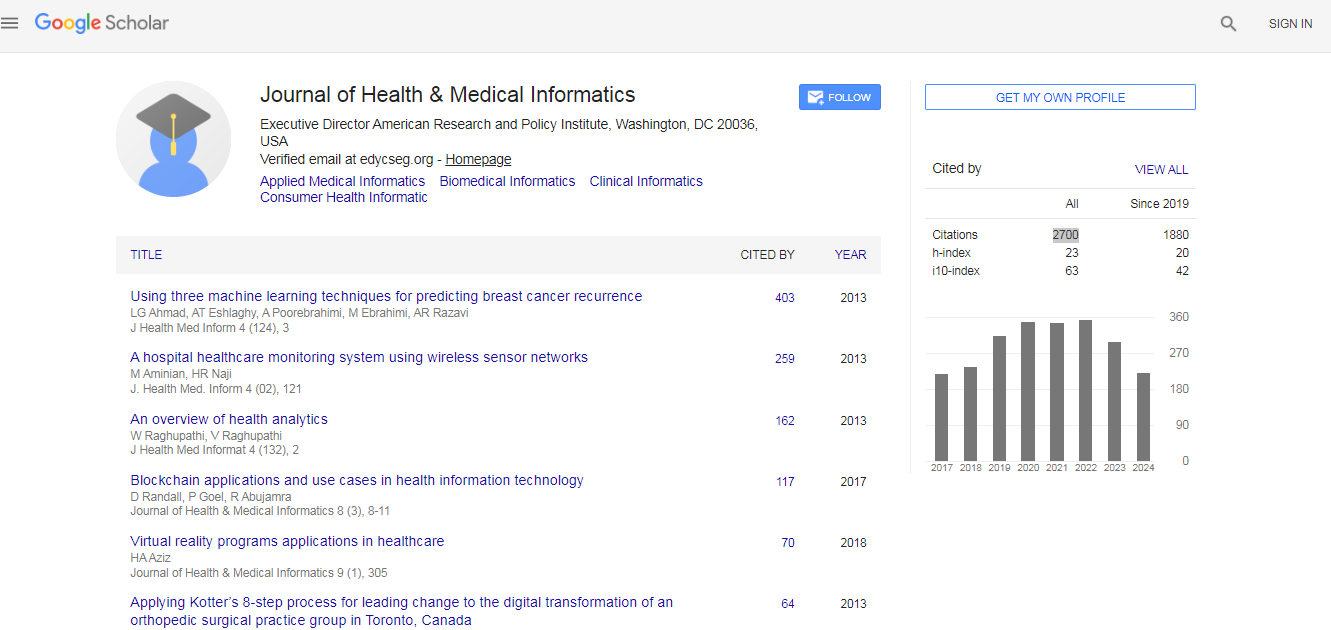
Journal of Health & Medical Informatics received 2700 citations as per Google Scholar report