Editorial - (2021) Volume 11, Issue 10
Received: 07-Oct-2021
Published:
17-Oct-2021
, DOI: 10.37421/2161-105X.2021.11.569
Citation: Lee, Zing. "An Overview on Interstitial Lung Diseases and Its Management." J Pulm Respir Med 11 (2021): 569.
Copyright: © 2021 Lee Z. This is an open-access article distributed under the terms of the creative commons attribution license which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Interstitial Lung Diseases (ILDs) are one of the most devastating consequences of systemic rheumatic disorders, causing major morbidity and mortality; they can also be the initial symptom of Connective Tissue Diseases (CTDs). The goal of this narrative review is to summarise the facts on CTD/ ILD pathophysiology and distinguishing aspects in various rheumatic diseases. The pathophysiology, clinical features, and therapy of ILD linked with rheumatic systemic disorders and CTDs were investigated using a combination of phrases or concepts to search the PubMed, Medline, and Cochrane Library databases for studies published between 1995 and February 2017 articles that are not. Expert opinions: Because of the paucity of reliable data on the treatments employed, the variability of the diseases itself, and the shortage of well-defined outcome metrics, managing CTD-ILD is difficult. Clinical treatment options are frequently determined based on functional and radiographic progression, as well as exacerbating factors including age and the weight of comorbidities. The management of CTD patients with ILD in CTD-ILD clinics necessitates interdisciplinary teamwork between rheumatologists and pulmonologists due to the complexity of diagnosis and the scarcity of therapeutic studies.
Interstitial Lung Disease (ILD) classification involves a multidisciplinary approach that includes input from a seasoned respirologist, a chest radiologist, and a lung pathologist. Despite a complete multidisciplinary review, up to 15% of ILD patients have unclassifiable ILD that cannot be diagnosed. However, because to a lack of research in this group, there are variations in the classification and terminology of unclassifiable ILD. Additional research is needed to discover the best way to assess and manage patients with unclassifiable ILD. Interstitial lung diseases are a group of diffuse parenchymal lung diseases with high morbidity and mortality rates. Recent advances in knowledge have resulted in the publishing of a new categorization of idiopathic interstitial pneumonias, which divides the disease into three categories: significant, rare, and unclassified. The new classification is unique in that it allows difficult-to-classify things to be treated using the disease behaviour classification. Idiopathic pulmonary fibrosis is the most fatal of the interstitial lung diseases, with a wide range of clinical manifestations. In clinical trials, a variety of biomarkers have been proposed to predict the course of the disease and to group patients with similar characteristics. In the field of other interstitial lung diseases, early diagnosis and disease stratification are also critical.
The lung is a common site of systemic Connective Tissue Disease (CTD) consequences, and lung involvement can manifest in a variety of ways. The most common lung manifestations of CTD are Interstitial Lung Disease (ILD) and pulmonary hypertension. Although it is commonly assumed that interstitial lung disease emerges later in CTD, it is frequently the first symptom ("lung dominant" CTD). Rheumatoid arthritis, scleroderma, systemic lupus erythematosus, polymyositis or dermatomyositis, Sjögren's syndrome, and mixed connective tissue disease are all examples of CTD with ILD. Despite parallels in clinical and pathologic appearance, CTD related ILD (CTD-ILD) has a very different prognosis and therapy than other types of ILD, such as idiopathic pulmonary fibrosis. Pulmonary hypertension (PH) can occur as a primary vasculopathy in pulmonary arterial hypertension or in the presence of ILD (PH-ILD). To diagnosis CTD-ILD, a complete history, physical examination, targeted serologic tests, and, on rare occasions, lung biopsy is required, whereas to identify pulmonary hypertension, both non-invasive and invasive assessments of pulmonary hemodynamics are required.
Immunosuppression is the most common treatment for ILD, while there is a dearth of data from Randomised Controlled Trials (RCTs) to back up specific treatments. Furthermore, treatment options fluctuate depending on the clinical circumstances; for example, a patient freshly diagnosed with CTD-ILD receives different treatment than someone experiencing an acute exacerbation of the disease. Immunosuppression is utilised only in rare cases of pulmonary arterial hypertension caused by CTD; most patients are treated with selective pulmonary vasodilators. Comorbidities such as sleep disturbed breathing, dyspnea symptoms, and cough should be assessed and treated for both conditions. In patients with severe disease, lung transplantation should be explored, however it is not always possible due to other CTD manifestations and comorbidities. To determine the optimal treatment techniques, clinical trials of novel medicines, including immunosuppressive therapies, are required.
Patients with RA-related ILD are also at a higher risk of infection and drug toxicity, which, along with comorbidities, makes treatment decisions even more difficult. Various clinical phenotypes, natural histories, and prognoses are associated with different histopathologic patterns of RA-related ILD. Idiopathic emphysema, the most common and severe of the idiopathic interstitial pneumonias, shares a number of clinical and histologic features with the Usual Interstitial Pneumonia (UIP) subtype of RA-related ILD, suggesting the presence of common mechanistic pathways and possibly therapeutic targets. There are still significant gaps in our understanding of RA-related ILD. Expert centres working together to clarify the basic mechanisms underlying RArelated UIP and other subtypes of RA-related ILD has the potential to speed the development of more effective and safer medications.
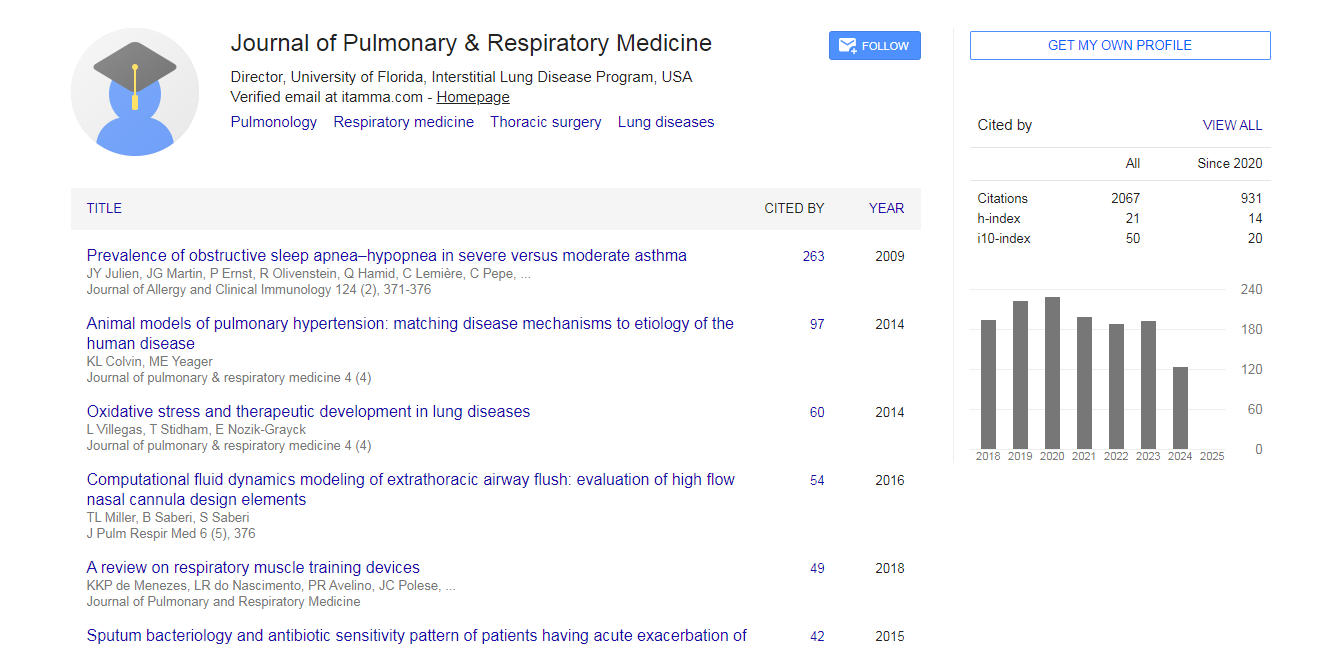
Pulmonary & Respiratory Medicine received 1690 citations as per Google Scholar report