Perspective - (2022) Volume 8, Issue 3
Received: 02-May-2022, Manuscript No. jotr-22-72061;
Editor assigned: 04-May-2022, Pre QC No. P-72061;
Reviewed: 18-May-2022, QC No. Q-72061;
Revised: 23-May-2022, Manuscript No. R-72061;
Published:
30-May-2022
, DOI: 10.37421/2476-2261.2022.8.202
Citation: Ghosh, Harish. “Combining Methods to Decode the Epigenome for Cancer Research and Diagnostics is Key.” J Oncol Transl Res 8 (2022): 202.
Copyright: © 2022 Ghosh M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Genome guideline is represented by the elements of chromatin alterations. The broad and different cluster of DNA and histone changes permit various components to act combinatorically and direct tissue-explicit and cellexplicit results. However, our capacity to clarify these mind boggling mixes and connection them to ordinary genome guideline, as well as comprehend their liberation in malignant growth, has been blocked by the absence of reasonable advancements. Here, we portray late discoveries demonstrating the significance of the combinatorial epigenome, and novel techniques to gauge and describe these mixes. These integral techniques range various disciplines, giving a way to unravel epigenetic mixes and connection them to organic results. At last, we examine the commitment of bridling the rich combinatorial epigenetic data to further develop malignant growth diagnostics and checking [1].
Cell-type explicit chromatin association, administered by record factors (TFs) and chromatin controllers, gives the means to different aggregates emerging from indistinguishable hereditary data. At the premise of the chromatin structure are the nucleosomes, in which the twofold abandoned DNA is folded over the histone octamer. Both the DNA and the four center histone proteins are broadly and powerfully changed by different covalent alterations, influencing various parts of chromatin homeostasis. Histone alterations direct chromatin pressing and 3D adaptation, accordingly controlling access of DNArestricting proteins. Of note, this capacity isn't confined to polar alterations like acetylation; mono-methylation on H3 lysine 20 was as of late displayed to manage chromatin collapsing. Moreover, methylation of H3 lysine 9 advances the arrangement of fluid stage division, accordingly directing chromatin compartmentalization and genome capability. At long last, various models exist for histone and DNA changes going about as extraordinary docking locales for the limiting or arrival of downstream effector proteins [2].
The significance of the epigenetic network is reflected in the imperative jobs it plays in cell separation and its repetitive liberation in malignant growth. While over half of human malignant growths harbor transformations in catalysts that are associated with chromatin association, the atomic components by which disease cells seize the epigenetic hardware changes between cancer types and firmly partner with the formative beginnings of the various cancers. Chromatin remodelers like the SWI/SNF complex, as well as focal histone modifiers like PRC1/2 and COMPASS, are repetitively dedirected in growths, bringing about significant changes to the epigenetic scene that upholds tumorigenic transcriptional programs. Repetitive transformations in the qualities encoding the center histones have likewise been connected to explicit growth. While the transformations show up either in the globular space or the widely altered histone tails, the subsequent freak histones, named 'oncohistones', are integrated into the nucleosome complex. Curiously, notwithstanding the low predominance of oncohistones in the genome, these transformations significantly influence the epigenome and transcriptome of these malignant growths. At long last, cancer cells get adjusted DNA methylation (5mC) and hydroxymethylation (5hmC) states because of direct transformations or modified movement of the DNA methylation apparatus, which might additionally modify the histone code and record, to keep up with the favorable to tumorigenic cell state [3,4].
The significance of the epigenetic network is reflected in the imperative jobs it plays in cell separation and its repetitive liberation in disease. While over half of human diseases harbor changes in catalysts that are engaged with chromatin association, the sub-atomic components by which malignant growth cells capture the epigenetic apparatus shifts between cancer types and firmly partner with the formative beginnings of the various growths. Chromatin remodelers like the SWI/SNF complex, as well as focal histone modifiers like PRC1/2 and COMPASS, are repetitively de-controlled in growths, Bringing about significant changes to the epigenetic scene that upholds tumorigenic transcriptional programs [5].
Repetitive changes in the qualities encoding the center histones have likewise been connected to explicit growths. While the changes show up either in the globular space or the broadly altered histone tails, the subsequent freak histones, named 'oncohistones', are integrated into the nucleosome complex. Strangely, in spite of the low predominance of oncohistones in the genome (because of numerous alleles encoding the wt histone), these changes significantly influence the epigenome and transcriptome of these tumors. At long last, cancer cells secure changed DNA methylation (5mC) and hydroxymethylation (5hmC) states because of direct transformations or modified movement of the DNA methylation hardware, which might additionally modify the histone code and record, to keep up with the favorable to tumorigenic cell state.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
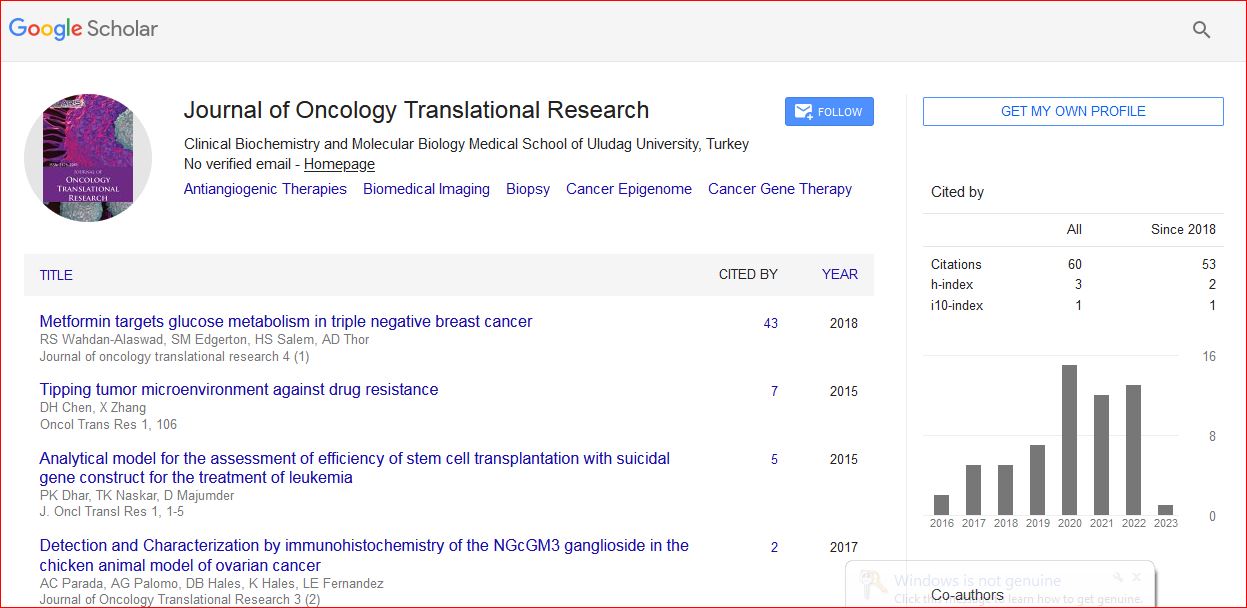
Journal of Oncology Translational Research received 93 citations as per Google Scholar report