Mini Review - (2022) Volume 11, Issue 12
Received: 25-Nov-2022, Manuscript No. jhoa-23-86322;
Editor assigned: 28-Nov-2022, Pre QC No. P-86322;
Reviewed: 13-Dec-2022, QC No. Q-86322;
Revised: 19-Dec-2022, Manuscript No. R-86322;
Published:
27-Dec-2022
, DOI: 10.37421/2167-1095.2022.11.381
Citation: Gaciong, Zbigniew. “Congenital Heart Disease and Chronic Pulmonary Hypertension.” J Hypertens 11 (2022): 381.
Copyright: © 2022 Gaciong Z. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Pulmonary hypertension is defined as a mean arterial pressure (MAP) of less than 25 mmHg. With an emphasis on their relevance to congenital heart disease, we investigate the pathogenesis, diagnosis, and treatment of the disease. Pulmonary hypertension is a fairly common complication of congenital heart disease, with an adult prevalence of 5 to 10%. The acknowledged multifactorial cause includes the size and type of the heart abnormality, as well as hereditary and environmental factors. More complex diseases are becoming more widely recognized in place of the pure Eisenmenger complex. A variety of tests, including echocardiography, exercise testing, cardiac catheterization, MRI, and CT scanning, can be used to identify increased pulmonary vascular resistance caused by remodelling of the pulmonary vascular bed. In management, disease-treating medications are utilized, and their efficacy is increasing.
Pulmonary hypertension • Eisenmenger • Pulmonary vascular resistance • Congenital heart disease
About 10% of adult cases of congenital heart disease (CHD) have a complication known as pulmonary hypertension (PH). It is characterised by a mean pulmonary arterial pressure (PAPm) of less than 25 mmHg, as measured at rest during right cardiac catheterization. Sufferers with precapillary PH are classified as having pulmonary arterial hypertension (PAH), a subgroup of PH patients. A pulmonary artery wedge pressure 15 mmHg and a pulmonary vascular resistance (PVR) >3 Wood Units in patients without any other precapillary PH causes, such as lung disease and persistent thromboembolic pulmonary hypertension, are indicative of this.
There are 97 cases of PH per million people in the UK, with a female-tomale ratio of 1.8, according to reports. In addition, the age-standardized PH death rate in the United States ranges from 4.5 to 12.3 per 100,000 people. PAH is found in 5–10% of adults with CHD. This has a significant impact on mortality and morbidity and raises the need for lifetime care. A Dutch study that looked at the epidemiology of septal abnormalities found that 6.1% of them had CHD-APAH, had a median age of 38, and 60% were women. The prevalence of CHD-APAH in the population is 15.6 per million, with Eisenmenger syndrome accounting for 58% of cases, and the estimated incidence is 2.2 per million. In addition, ventricular septal defect (VSD) was the most common underlying defect, accounting for 42% of cases. This study also emphasizes the risk of PH following surgery to fix seal defects, which results in 3% of these people still experiencing PH. The fact that the severity of CHD-APAH can vary significantly even in the presence of similar underlying cardiac abnormalities highlights the dynamic nature of the syndrome and its complex genesis [1]. The cause of CHD-APAH is determined by the underlying abnormality, but environmental factors, genetics, and/or epigenetics may also be involved. A study that found that BMPR2 mutations were found in 6% of people with CHDAPAH also have a less strong (75 and 25%, respectively) connection to familial and idiopathic PAH. This, as well as mutations in the gene that makes the protein ALK-1, are well known to cause PAH. Left heart obstructive disease and postcapillary hypertension are the two most common secondary causes of pulmonary hypertension in CHD patients. Common diseases include VSD, persistent ductus arteriosus, and atrial septal defect (ASD). Studies show that the severity of the abnormality influences whether patients develop PAH [2].
For instance, the natural course of VSD patients reveals that 3% of individuals with mild or moderate abnormalities (1.5 cm in diameter) will go on to develop Eisenmenger syndrome. However, if there is no surgery, the condition will eventually occur in all major abnormalities (>1.5 cm). A recent article for ASDs supports the same conclusion, showing that the greatest defects (31.84 8.21 mm) were those most likely to have severe PAH. Early PAH is frequently associated with more complicated abnormalities, such as atrioventricular septal defects or truncus arteriosus. Furthermore, once the underlying heart defect has been fixed, people may still be diagnosed with PAH [3]. It's unclear whether this is because the pulmonary vascular disease has progressed despite surgical correction, although research suggests that early correction works to delay the onset of PAH in the future. Particularly, after the fontal procedure, PH has attracted a lot of interest. This procedure is the last resort for children born with single-ventricle CHD who are receiving stepwise palliation. Over the past 20 years, there have been significant advancements in early outcomes due to early surgical success, but pulmonary hypertension is still associated with late mortality and morbidity. Reduced ventricular filling and subsequently reduced cardiac output are caused by the absence of a subpulmonary ventricle, which helps to pump blood through the pulmonary vasculature. Long-term PVR to blood flow via the pulmonary system increases, although pressures are not elevated over a mean of 25 mmHg [4].
Lack of pulsatile flow, poor respiratory mechanics, and diminished systemic ventricle diastolic, long axis, and systolic function are some of the factors that contribute to the distinct pathogenesis. Even though the lowpressure system does not immediately raise the PAP, the pulmonary circulation becomes volume-overloaded as a result. The primary factors that influence the likelihood of developing PAH are the size of the ASD and the compliance of the right ventricle. However, left cardiac lesions and left ventricle dysfunction may play a role. Only 2% of ASD patients develop Eisenmenger in pretricuspid lesions, indicating its rarity. Due to post-tricuspid lesions, high-pressure left-toright shunts overload the left ventricle and pulmonary circulation with volume. In some abnormalities, PAH begins within the first few years of life. If this is not addressed, the Eisenmenger complex, a type of super systemic PVR, will almost always develop, resulting in shunt reversal [5].
Vasoconstriction, medial wall hypertrophy, and pulmonary vascular bed remodelling are all factors in Eisenmenger's pathophysiology of PAH. The pulmonary arteries' histology in CHD-APAH reveals plexiform lesions, medial hypertrophy, intimal proliferative fibrosis, an expansion of smooth muscle cells into the peripheral pulmonary arteries, and rarefication of the pulmonary artery tree. High flow and pressure are thought to be the culprits behind the breakdown of endothelial barrier function, which in turn damages the pulmonary vascular endothelial cells. As a result, the extracellular matrix is broken down and FGF and TGF-1 are released, which activates vascular elastase and matrix metalloproteinase and causes them to work more efficiently. This release triggers the growth and proliferation of smooth muscle cells as well as the development of neo-intima. It is believed that inflammation and thrombosis result from the adhesion and activation of platelets and leukocytes following endothelial injury, with subsequent activation of the coagulation pathways. Overall, the endothelial dysfunction and subsequent pulmonary artery vascular remodelling cause elevated PVR and ultimately right ventricular failure.
Initial symptoms are frequently brought on by exertion and are connected to advancing right ventricular failure. There may be weariness, angina, syncope, and shortness of breath. With studies demonstrating that >90% of patients are in WHO class II or worse and 50% express severe limits, persons with Eisenmenger complex will typically have exercise intolerance. Symptoms can be felt when you're at rest in more severe cases. Furthermore, if right heart failure worsens, traditional symptoms such abdominal distension and ankle oedema may appear. Less frequently occurring symptoms include hoarseness caused by constriction of the left recurrent laryngeal nerve linked to dilated pulmonary artery and haemoptysis related to rupture of hypertrophied bronchial arteries. Angina occurs when the left major coronary artery is compressed between the dilated pulmonary artery and the aorta, whereas wheezing occurs when the big airways are compressed. Left parasternal lift, a loud second heart sound, a third heart sound connected to the right ventricle, pan systolic murmur (tricuspid regurgitation), and diastolic murmur are all clinical indicators of pulmonary hypertension (pulmonary regurgitation). Hepatomegaly, ascites, peripheral edoema, and cold peripheries may be present if PAH has progressed, as well as elevated jugular venous pressure. The appearance of clubbing, hepatic and renal failure, ischemic consequences, and the signs of endocarditis should all be noted because patients who are cyanosed frequently exhibit these symptoms.
Electrocardiography (ECG), chest radiography, peripheral oxygen saturations, objective assessment of exercise tolerance, and echocardiography are a few of the fundamental tests that should be performed first. The right ventricle and pulmonary vascular bed can then be studied further using chest computed tomography (CT) and magnetic resonance imaging (MRI). Electrocardiography it should be made clear that while an electrocardiogram can show signs of PH, a normal ECG does not rule out the diagnosis. Right ventricular strain and hypertrophy, right bundle branch block, right axis deviation, and right atrial hypertrophy are all possible CHD-APAH findings. The highest amplitude of the S wave in V5 or V6 and the R-wave amplitude in V1 can be added to determine the latter. There is evidence that this could offer specific predictive information for Eisenmenger complex patients. RV strain is more sensitive than RV hypertrophy in demonstrating PH, according to studies. The mean frontal QRS axis may be rightward even under normal circumstances in patients with complicated CHD, which is especially true. X-rays of the chest Pulmonary artery dilatation, aneurysms, or calcification are chest radiographic findings in CHD-APAH patients. The cardiothoracic ratio can be determined, and there may also be right atrial and/or right ventricular hypertrophy. Pulmonary venous congestion is a symptom of left heart disease. Additionally, consolidation brought on by infiltrates or pulmonary bleeding may occur. Despite this, many radiographs taken of these patients may be normal. testing of exercise This can be accomplished through either a 6-minute walk test or cardiopulmonary exercise testing with peak oxygen consumption monitoring. Both are frequently used to measure CHD-APAH, and both a decrease in distance in the former and a decrease in peak oxygen in the latter have been linked to a worse prognosis in these patients. Additionally, evidence of desaturation during exercise suggests that the shunt may be reversing, and evidence of probable right-to-left shunting in ASDs suggests a likely increased PVR and aids in the prediction of survival.
ICHD-APAH should be treated in specialised centres that see CHD and PH patients on a regular basis. A number of reviews in the UK and other countries are helping to clarify the standards that apply to CHD-PAH centres. Furthermore, patient education, awareness of potential risks and complications, and behavioural changes are critical in the care of these patients. Strenuous exercise is discouraged, but light activities are beneficial. Patients can experience clinical deterioration at any time during their treatment. These include dehydration, lung infections, high altitudes, and noncardiac surgery requiring general anaesthesia. Since pregnancy carries a high risk for both the mother and the foetus, effective contraception is crucial. Endothelia receptor antagonist users should use dual contraception due to the interaction of contraceptives with progesterone-based substances.
Diuretics are typically used for any fluid build-up during CHD-APAH therapy. Treatment for hepatic congestion, ascites, and peripheral oedema is symptomatic because this is a late manifestation. In real life, arrhythmias are the only situation where digoxin is employed. There is relatively little research supporting the use of oxygen treatment, which is basically only indicated for patients who have low night-time oxygen saturation levels, particularly in the presence of airway obstruction or coexisting long illness. Anticoagulants and calcium channel blockers are not recommended in CHD. Anticoagulation treatment specifically increases haemoptysis-related mortality in CHD-APAH patients. Calcium channel blockers that do not contain nondihydropyrodine are not advised since they are negatively inotropic.
Given the reductions in morbidity and mortality associated with early surgical intervention, it is reasonable to anticipate earlier detection and treatment of CHD-APAH through ongoing education and research. It is unknown whether this will lower the prevalence of the condition. Studies currently being conducted into the treatment of PH, particularly in patients with CHD [3,4,6], may lead to future enhancements in survival rates. For instance, trials for the novel dual receptor antagonist macitentan are currently being carried out in order to assess whether or not it is effective in treating the fundamental form of Eisenmenger syndrome. The majority of people tolerate it well, and oxygen saturation and efficiency appear to be improving. A better understanding of the disease processes associated with PH and CHD could lead to potential treatments in the future. Additionally, the underlying pathophysiology of the effects on lung function is still poorly understood.
None.
None.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
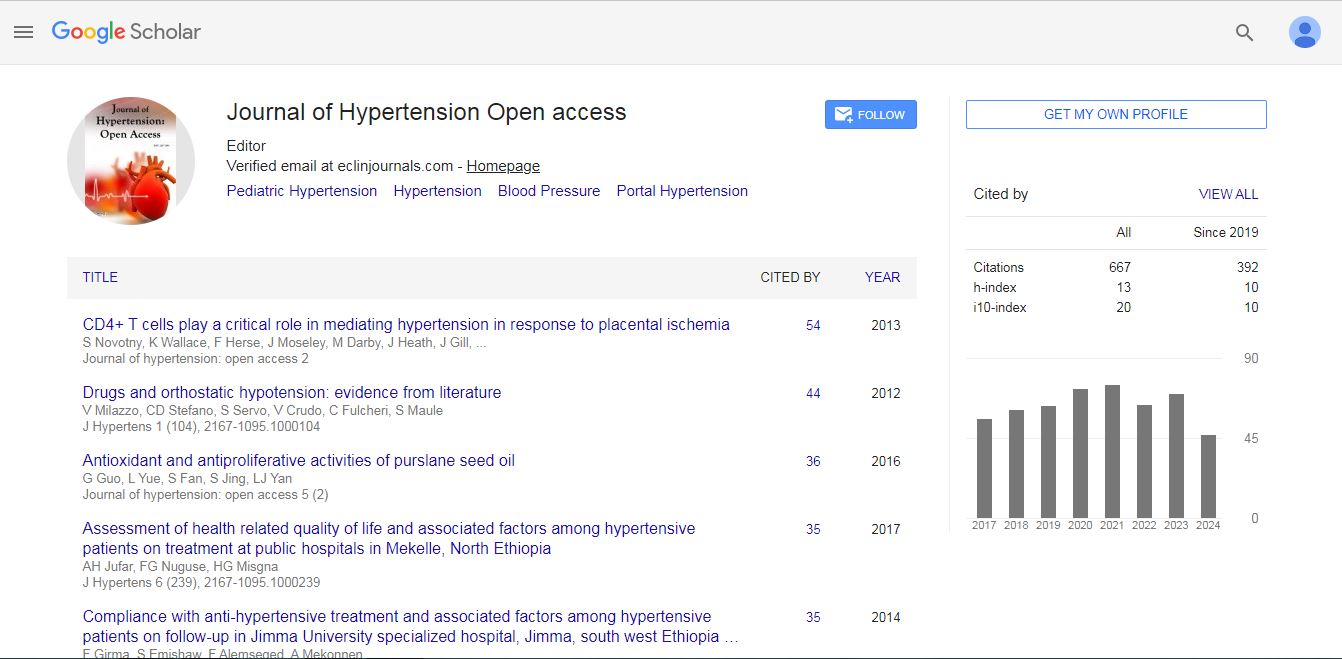
Journal of Hypertension: Open Access received 614 citations as per Google Scholar report