Opinion - (2023) Volume 8, Issue 1
Received: 02-Jan-2023, Manuscript No. Jcct-23-96422;
Editor assigned: 04-Jan-2023, Pre QC No. P-96422;
Reviewed: 16-Jan-2023, QC No. Q-96422;
Revised: 21-Jan-2023, Manuscript No. R-96422;
Published:
28-Jan-2023
, DOI: 10.37421/2577-0535.2023.8.204
Citation: Friedmann, Theodore. “Cytosolic DNA Sensors in the
Tumour Microenvironment Regulate Immunity.” J Cancer Clin Trials 8 (2023): 204.
Copyright: © 2023 Friedmann T. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
cGAS and AIM2 are CDSs that are expressed in a variety of cell types, including immune and tumor cells and are activated when cytosolic dsDNA is present. Anti-tumor immune responses are enhanced when tumor-derived dsDNA is recognized by CDSs in the cytosol of tumor-infiltrating dendritic cells (TIDCs). This activates both innate and acquired immunity. STING is the cGAS effector that triggers type I interferon (IFN) signaling in the downstream signaling pathway. STING agonists have been injected intratumorally in a number of clinical trials to boost the anti-tumor immune response elicited by immune checkpoint antibodies due to their ability to activate TIDCs. But they haven't done much, which shows how important it is to get the right dose and route of STING agonists and figure out other immune pathways that help immune responses fight tumours. Recent research has shown that multiple parallel pathways, including the inhibition of STING-type I IFN signaling, are involved in the pro-tumour growth induced by AIM2 activity. As a result, cancer immunotherapies might want to target AIM2 on a molecular level.
This review provides a synopsis of the most recent research on the roles that cGAS, STING and AIM2 play in immune cells as well as tumor cells in the tumor microenvironment. It also discusses the potential for anti-tumor treatment strategies that are based on these molecules in the near future. Majority of studies focus on tumor-resident immune cells. As a result, there are not many data on how STING works in tumor cells. Concentrates on utilizing mouse and human colon malignant growth tests, as well as cellular breakdown in the lungs model mice and the TCGA cellular breakdown in the lungs dataset, found that cancer cells smother the significant level articulation of cGAS and STING through epigenetic guideline or the creation of DNase. The following novel treatment strategy was attempted in light of these characteristics: Cyclic dinucleotides (CDNs) clung to STING (which had been modified to forestall debasement by DNase) were brought into the growth of a melanoma mouse model. After that, chemotherapy and radiation therapy induced apoptosis artificially, releasing a large number of CDNs into the tumor's microenvironment. This demonstrated that CDNs in combination with radiation or chemotherapy could be an innovative treatment option for activating STING in TIDCs and had a significant effect on activating the STING type I IFN signaling pathway [1].
Some types of cancer have shown signs of chromosomal instability (CIN), in which some or all of the chromosomes change numbers. cGAS and dsDNA are more likely to bind in cancers with a "CIN-high" score than in cancers with a "CIN-low" score. There are discrepancies, such as a high likelihood of cancer cell proliferation and metastasi despite the fact that STING is activated and the type I IFN signaling pathway that follows it. In CIN-high malignant growths, IRF3 and NF-κB1 don't actuate the sort I IFN flagging pathway downstream of STING. Instead, activation of noncanonical NF-B (NF-B2) signaling suppresses this signaling pathway. Despite the activation of tumor cell STING signals, this finding suggests that the activation of the type I IFN signaling pathway may be suppressed. In this way, the therapy ought to prompt the arrival of cGAMP and CDNs from cancer cells into the growth's microenvironment to actuate STING in the cDC1 TIDCs that have penetrated the carcinogenic development, consequently improving the counter cancer resistant reaction [2].
As a possible adjuvant for increasing the antitumor immune response elicited by anti-PD-1 immunotherapy, STING agonists that activate cDC1 TIDCs have been investigated. However, the administration of intratumoral STING agonists and anti-PD-1 antibodies as part of a treatment regimen has not produced encouraging outcomes. In this manner, STING agonists that can be managed fundamentally and balance out the nearby affirmation of STING to diminish the harmfulness might be a compelling methodology for focusing on the STING-type I IFN flagging pathway. In future clinical trials, anti-PD-1 Ab+ systemic STING agonist combination therapy should be investigated because, once optimized, it has the potential to alter the therapeutic landscape [3].
What's more, there stays a need to break down other insusceptible pathways that add to the invulnerable penetration into cancers and subsequently help to decide their hot or cold states. Antibodies against TIM-3, an immune checkpoint receptor that regulates CD8+ T-cell function and inhibits the effectiveness of STING agonists, have been tested by researchers. It would be impossible to develop a treatment that targets AIM2 in the near future because there is no drug that inhibits it specifically. Anti-AIM2 therapy would activate TIDCs and be an IL-1 and IL-18 inhibitor in addition to being used as a STING agonist, thereby affecting more pathways than the STING agonist does [4,5].
As a result, AIM2 might be able to strengthen antitumor immunotherapies more effectively as a candidate for molecularly targeted drug therapy. The engineering of a nano molecular STING agonist vaccine that has been modified to be taken up by cDC1 has recently been reported by researchers. The immunotherapeutic antitumor effect of the anti-PD-1 Ab may be enhanced by an AIM2 siRNA nano vaccine made with the same technology. Antitumor immunotherapies are also expected to benefit greatly from AIM2 inhibition in TIDCs, Tregs and macrophages. As a result, a novel cancer immunotherapy that targets CD45+ cells specifically could be an AIM2 siRNA nano vaccine.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
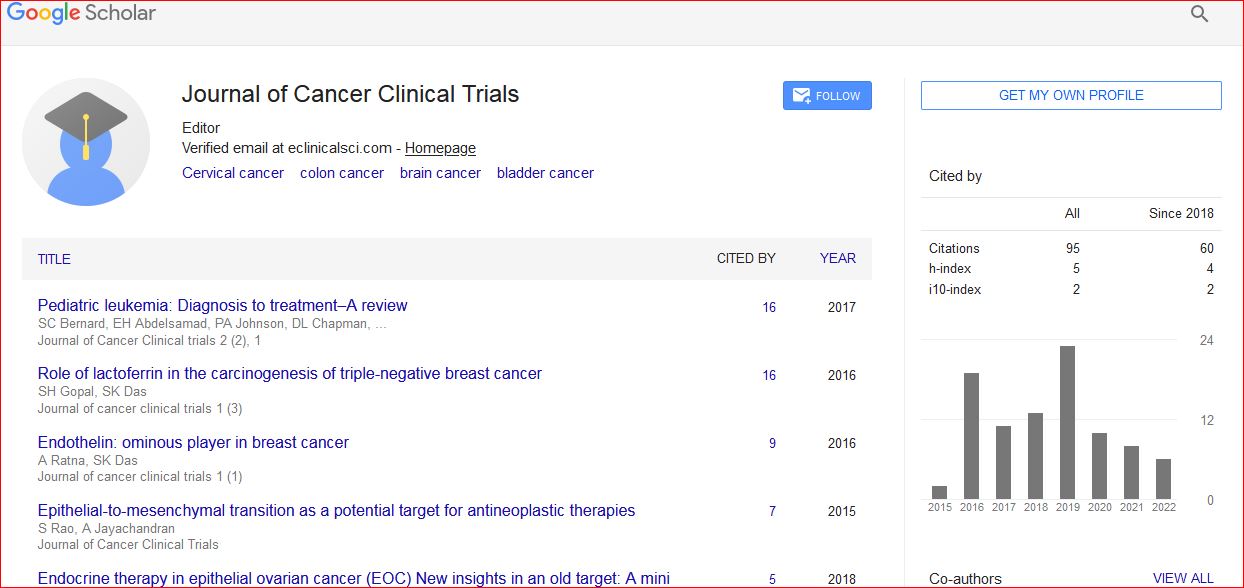
Journal of Cancer Clinical Trials received 95 citations as per Google Scholar report