Case Report - (2022) Volume 14, Issue 5
Received: 10-May-2022, Manuscript No. jcst-22-63645;
Editor assigned: 11-May-2022, Pre QC No. P-63645;
Reviewed: 21-May-2022, QC No. Q-63645;
Revised: 24-May-2022, Manuscript No. R-63645;
Published:
31-May-2022
, DOI: 10.37421/1948-5956.22.14.529
Citation: Lobato-Polo, Javier. “Erdheim-Chester Disease with a Posterior Fossa Tumour Mimicking Neurosarcoidosis: A Case Report.” J Cancer Sci Ther 14 (2022): 529.
Copyright: © 2022 Lobato-Polo J. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Erdheim-Chester Disease (ECD) is a rare non-Langerhans' cell histiocytosis described in 1930 by Jakob Erdheim and William Chester, it can present as a multisystemic entity that forms xanthogranulomas which are foamy histiocytes surrounded by fibrotic tissue. Lesions are commonly located in long bones, Central Nervous System (CNS), cardiovascular system, lungs, kidneys and skin. The CNS is involved in approximately 50% of cases and can compromise both extra and intra-axial structures and therefore can mimic schwannomas or meningiomas, amongst other mass lesions. Clinical presentation will differ from patient to patient thus diagnosis depends greatly in imaging, immunohistochemistry and genetic findings within the pathology analysis. The pathogenesis of this disease remains unknown. It is most commonly found in the middle-aged male population. Here, we present a case of a middle-aged woman with an extra-axial lesion that was initially considered to be neurosarcoidosis proving the diagnostic challenge this entity implies.
Erdheim-Chester disease • Posterior fossa tumour • Histiocytosis • Skull base • Case report
ECD: Erdheim-Chester Disease; CNS: Central Nervous System; MRI: Magnetic Resonance Imaging; CBC: Complete Blood Count.
A 54-year-old female with a past medical history of systemic arterial hypertension, presented with hemicrania occipital oppressive headache, associated with numbness in both arms and legs, and progressive gait instability. Her initial evaluation was elsewhere and included magnetic resonance imagining (MRI) that reported a mass lesion in the inferior clival region surrounding the right vertebral artery. Embolization of the right vertebral artery was performed, and the patient was referred to the out-patient clinic. She would reach our hospital three months later.
Upon examination, she presented mild dysarthria, deviation of the tongue to the right, diminished strength in all four extremities and truncal ataxia. A new brain MRI was performed (Figure 1). Other nodular lesions were observed in both orbits, lacrimal glands and in retromaxillary areas and pterygopalatine fossa. Neurosarcoidosis was the working diagnosis at this stage. All blood tests were normal but one slight elevation in the Reactive-C Protein (RCP). A chest X-Ray was performed to rule out systemic sarcoidosis and was reported as normal [1-9].

Figure 1.Brain MRI. (A) Axial T2-weighted sequences show a right paramedian mass in the inferior third of the clivus with extension through the foramen magnum, with some hypointense spicules emanating from the centre of the mass, surrounding the right vertebral artery, (B) sagittal T1-weighted sequences show a compressing mass of the pons and medulla with hyperintense areas corresponding to previous endovascular treatment, (C) sagittal T1-weighted post gadolinium image of a right clival paramedian extra-axial mass demonstrates necrosis areas and avid contrast enhancement, (D) axial T1-weighted post gadolinium, (E) postsurgical MRI. Axial T1-weighted post gadolinium image shows the decreased size and compressive effect over the pons and (F) axial T2-weighted shows postsurgical changes.
The patient underwent a right retromastoid craniotomy. We used ultrasonic surgical aspirator for tumour debulking. No cranial nerves were visibly lesioned.
Following the surgery, in the intensive care unit, the asymmetric elevation of the soft palate was detected as well as peripheral facial palsy, House Brackman III. Later on, she was diagnosed with diabetes insipidus, rare given the mass location. Postoperative Computed Tomography (CT) scan showed no evidence of haemorrhage or acute hydrocephalus.
Pathology reported a xanthogranulomatous lesion without evidence of malignancy. Immunohistochemistry showed focal reactivity to S100, foamy histiocytic infiltrates positive for CD68 and negative for CD1a as well as CD45, CD3 and CD20 lymphocytic infiltrates. Neurosarcoidosis was still being considered, also the possibility of mycobacterial infection was contemplated given our regional epidemiology and IgG4 disease. The ratio of plasma cells IgG4/IgG was not greater than 40% not meeting diagnosis criteria. Chest CT showed a nodular pericardial thickening with periaortic inflammatory tissue indicative of periaortitis, pericardial and pleural effusion, interstitial pulmonary infiltrates, and sclerotic bone lesions. Erdheim-Chester disease was suggested at this point and later confirmed by bone scintigraphy and genetic screening where a mutation in the BRAF V600E/V600E2/V600D was detected and oriented treatment (Figures 2-5).

Figure 2. A xanthogranulomatous inflammatory process was evidenced with areas of ischemic necrosis, without atypia or mitotic activity (A), Histiocytic cells were positive for CD68 (B), negative for CD1a (C) and Langherin (D). Also, mononuclear inflammatory cells are evident: T lymphocytes and B lymphocytes, positive for CD3 (E) and CD20 (F), respectively. (G) and (H) orbits MRI. Axial T1-weighted post gadolinium image shows bilateral Intra- and extraconal nodular lesions with avid contrast enhancement with bilateral optic nerve compression, the dimensions are 16x14 mm on the right side and 13 × 16 mm on the left side. (I) and (J) Bone scintigraphy demonstrates multiple areas of abnormally increased uptake especially in the appendicular skeleton, highlighting the symmetrical hypermetabolism of the humerus, ulna, radius, scapula, femurs, tibiae and calcaneus.

Figure 3. Western blot demonstrating increased HDAC-6 expression in both SJSA-1 and MCF-7 DoxR cells compared to their WT counterparts (A), as well as increased HIF-1α expression in DoxR cells compared to WT which is reduced with siRNA knockdown (D) and vorinostat (B). HDAC-6 precipitates with HIF-1α in DoxR but not WT SJSA-1 cells or DoxR cells treated with Vorinostat (C).

Figure 4.Matrigel in vitro invasion assay (mean and standard error) demonstrating histone deacetylase (HDAC) inhibition with vorinostat and small interfering RNA reduced the invasiveness of doxorubicin-resistant (DoxR) SJSA-1 (A) and MCF-7 cells (B) relative to their parental wildtype (WT) cells, (*) indication of significance (P < 0.05).

Figure 5. Small hairpin RNA (shRNA)-mediated inhibition of hypoxia inducible factor-1α (HIF-1α) reduced the invasiveness of doxorubicin-resistant (DoxR) cells. Western blot demonstrated successful shRNA knockdown of HIF-1α expression (A). Small hairpin RNA-mediated knockdown of HIF-1α reduced the invasiveness of SJSA-1 DoxR cells in the Matrigel in vitro invasion assay (B), (*) indication of significance (P < 0.05). Invasion was calculated as the percentage of cells able to invade through a membrane coated with Matrigel during a 24 h period as a fraction of the control. Bars represent the normalized invasion indices (mean ± standard error). Light microscopy (2.5X) images of the Hema 3-stained Matrigel membranes demonstrated the increased number of SJSA-1 DoxR cells able to invade the Matrigel matrix compared to the parental wild-type (WT) cells (C). The drastic reduction in the invasiveness of DoxR cells with HIF-1α shRNA knockdown is apparent.
Treatment with Alpha 2A interferon was considered but due to unavailability in the country, Vemurafenib was started with appropriate tolerance reported by the patient. As far as the writing of this report, the patient has received 6 months of therapy without side effects. Her dysphagia and facial palsy had improved, and the patient scored 3 in the modified Rankin Scale.
ECD is a rare non-Langerhans cell histiocytosis that causes a multisystemic disease that can compromise long bones with painful osteoclastic lesions, and can also involve retroperitoneum, cardiovascular, pulmonary or orbits and cause periaortitis, pericarditis, retroperitoneal fibrosis, pulmonary infiltrates or xanthelasmas [1,8]. Its CNS involvement is relatively common, around 50%, and can mimic various intra and extra-axial lesions generating a widespread of neurological manifestations and/or complications such as diabetes insipidus and hydrocephalus [6,9]. Its diagnosis is achieved by pathological examination, imaging findings and genetic studies.
On the other hand, neurosarcoidosis is an uncommon manifestation of sarcoidosis seen in about 5-35% of patients with sarcoidosis, occasionally presented in isolation without signs of systemic illness. It’s CNS compromise includes meningeal, cranial nerves, hypothalamus, spinal cord or peripheral nerve involvement showing granulomatous infiltrates in the pathology [7]. Its diagnosis requires all clinical histological, imaging, and laboratory test support, and given its broad clinical spectrum, it is considered one of the great imitators, masquerading itself as other diseases becoming a challenging diagnosis to come by, as well as a powerful confusing factor as seen in our case, initially considered to be a posterior fossa tumour.
The MRI showed that the posterior fossa lesion was isointense in those T1 and T2-weighted images with high contrast enhancement, and the orbit lesions were hypointense in the same sequences, this finding it’s not specific for neither of the conditions mentioned above. Both present with a meningeal enhancement and hyperintense lesions on the FLAIR sequences. For neurosarcoidosis the imaging findings can be classified according to Christoforidis GA, et al. [3] in six categories, being: (1) dural thickening or mass, (2) leptomeningeal involvement, (3) enhancing brain parenchymal lesion, (4) non enhancing brain parenchymal lesion, (5) cranial nerve involvement and (6) spinal cord and root involvement. In our case the patient did not fit just one of the categories mentioned above given that her lesion had both dural thickening or mass and cranial nerve involvement.
No such classification exists for ECD but given the cases reported in literature it could be divided according to the location of the lesions: (1) meningeal involvement with thickening and/or mass, (2) hypothalamicpituitary axis mass, (3) supratentorial brain parenchymal lesion excluding hypothalamic-pituitary axis, (4) infratentorial parenchymal mass lesions and, (5) cerebrovascular involvement with peri-arterial infiltration. Taking this into account, our patient showed meningeal involvement with a mass lesion, that is seen in 23% of ECD cases, and cerebrovascular involvement with orbital lesions [10]. The concomitant presentation with intraconal orbital lesions that are seen hypointense in the T2-weighted sequences are indicative of ECD and should expedite diagnosis as does the coexistence of neurologic symptoms that are not attributed to the intracranial lesion such as diabetes insipidus or cerebellar symptoms [8].
Initial testing did not include CSF analysis given that the patient presented itself with a posterior fossa tumour and the risk of the performing a lumbar puncture was high, therefore omitted. Pathology studies of both diseases show inflammatory changes within the tissue with avid lymphocytic infiltration and granulomatous lesions formation. Differentiation between both of them can be challenging and not even immunohistochemistry can distinguish them. For ECD, granulomas present with positive CD68 and negative CD1a, 20% of cases can present positive S100 which correlates with what’s seen in the patients’ pathology, but it also presents lymphocytic infiltrates of CD3 T-lymphocytes and B-lymphocytes positive for CD20 that is present in neurosarcoidosis [2,5]. The lack of Birbeck granules in the electronic microscopy could have helped provide an early diagnosis of ECD, but it was not analysed in our case due to the lack of this resource in our institution [9].
At this stage, the diagnosis of ECD was obscured by the possibility of neurosarcoidosis given that both diseases can present similar clinical manifestations, imaging findings on MRI and even undifferentiated histopathological infiltrates. Other differential diagnoses that could be taken into consideration are Langerhans cell histiocytosis, Rosai-Dorfman disease, juvenile xanthogranuloma, multiple sclerosis and granulomatous infectious diseases like tuberculosis, toxoplasmosis and fungal infections. Since tuberculosis was considered in our case a Chest CT was obtained giving the diagnostic possibility of ECD. It did not only show the presence of sclerotic lesions but also of periaortitis, pulmonary infiltrates and pericardial and pleural effusion. The confirmation was made by bone scintigraphy in which bilateral symmetric metadiaphyseal medullary sclerosis with epiphyseal sparring and intense uptake of the radiotracer is seen in long bones [1]. Cranial vault involvement has been reported in up to 26% of cases [6].
First-line treatment is considered to be interferon alfa for disease stabilization but given that this medication is not available in Colombia we were forced to consider second-line treatments. Amongst those are anakinra, vemurafenib, imatinib and cladarbine. Our treatment was directed by genetic studies that revealed positive for BRAF V600E proto-oncogene that suggest clonal proliferation giving the possibility of treatment with Vemurafinib and therefore inhibiting the activation of the RAS-extracellular-signal-regulated kinase limiting proliferation and lesion survival [1]. This mutation is present in 55-65% of ECD cases [10]. So far, the patient responded adequately to the treatment and no salvage therapies like vincristine, vinblastine, cyclophosphamide, and methotrexate have been used. Nowadays patients have a 1-year survival rate of 96% and 5-year survival rate of 68% [9].
This case is relevant so that it showcases the diagnosis difficulty encountered and the fact that some diseases or great imitators such as neurosarcoidosis or tuberculosis can lead to a delay in diagnosis and treatment for a patient with ECD. The presentation of an intracranial lesion accompanied by intraconal orbital tumours and lacrimal gland infiltrates must be carefully studied and ECD should be considered a possibility from the start and chest scans and bone scintigraphy is recommended early on.
We confirm that we have read the position statement of the journal regarding the issues involved in ethical publication and affirm that this work adheres to those guidelines. Written informed consent was obtained from the patient for the publication of this article and accompanying images as well as ethics committee approval.
This report was approved by the committee for ethical research of the Fundación Valle del Lili and written consent for its publication was granted by the patient.
No funding was secured for this study.
The authors declare there are no financial relationships relevant to this article.
The authors declare no conflicts of interest.
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
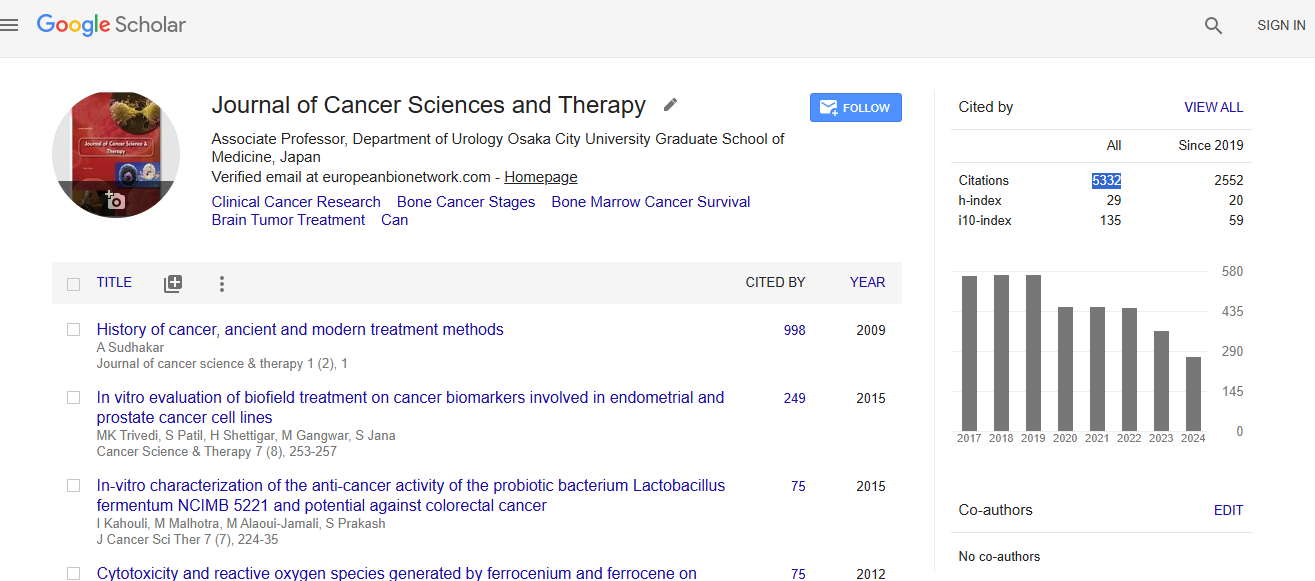
Cancer Science & Therapy received 5332 citations as per Google Scholar report