Commentary - (2024) Volume 14, Issue 4
Received: 02-Jul-2024, Manuscript No. jcde-24-143404;
Editor assigned: 04-Jul-2024, Pre QC No. P-143404;
Reviewed: 16-Jul-2024, QC No. Q-143404;
Revised: 22-Jul-2024, Manuscript No. R-143404;
Published:
29-Jul-2024
, DOI: 10.37421/2165-784X.2024.14.558
Citation: Chen, Jiehong. “First-principles Analysis of Band Structures and Transport Properties in High-performance Half-heusler Thermoelectric Materials.” J Civil Environ Eng 14 (2024): 558.
Copyright: © 2024 Chen J. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Thermoelectric materials, which convert temperature differences directly into electrical voltage or vice versa, have garnered significant interest due to their potential in waste heat recovery and solid-state cooling applications. Among the myriad materials studied for these purposes, Half-Heusler compounds stand out due to their promising thermoelectric properties and high performance. A Half-Heusler alloy is a type of intermetallic compound with a specific crystallographic structure that can be described by the general formula XYZ, where X, Y and Z are different elements. These materials are characterized by their unique combination of electrical and thermal properties, making them suitable candidates for thermoelectric applications. To optimize the performance of thermoelectric materials, a detailed understanding of their electronic structure and transport properties is crucial. This is where first-principles calculations, based on quantum mechanical principles, come into play. First-principles analysis allows for the investigation of material properties without empirical parameters, relying solely on fundamental physical constants and the principles of quantum mechanics. This method provides insights into the band structure and transport properties of materials, which are essential for predicting their thermoelectric performance. Half- Heusler alloys, due to their tunable band structures and diverse elemental combinations, offer a rich field for exploration. Their band structures often feature narrow band gaps or semi-metallic behaviors that can be finely tuned through composition and structural modifications. Understanding these band structures is crucial as they directly influence the electronic density of states, which in turn affects the thermoelectric efficiency of the material. In this comprehensive analysis, we will explore the first-principles methods used to investigate the band structures and transport properties of high-performance Half-Heusler thermoelectric materials. We will delve into the computational techniques employed, the theoretical frameworks applied and the implications of these analyses for material design and optimization [1].
First-principles calculations, often referred to as ab initio calculations, are computational methods used to study material properties based solely on fundamental physical principles, without relying on empirical data. The cornerstone of these methods is Density Functional Theory (DFT), which provides a framework for calculating the electronic structure of materials. DFT solves the Schrödinger equation for the electrons in a material, allowing us to obtain the band structure, density of states and other electronic properties. For Half-Heusler compounds, DFT calculations begin with the construction of a supercell representing the material's crystal structure. The exchangecorrelation potential in DFT is typically approximated using various functionals, such as the Local Density Approximation (LDA) or the Generalized Gradient Approximation (GGA). These functionals account for the complex interactions between electrons and provide a starting point for further analysis [2].
In addition to DFT, other first-principles methods, such as the GW approximation and Dynamical Mean-Field Theory (DMFT), can be employed to refine the electronic structure calculations. The GW approximation corrects for the shortcomings of DFT by providing a more accurate description of the quasiparticle energies, while DMFT addresses the strong electron-electron correlations present in some materials. The band structure of a material describes the allowed and forbidden energy levels of electrons as a function of their momentum. For Half-Heusler alloys, the band structure is influenced by the specific elements involved and their arrangement in the crystal lattice. The band structure can reveal crucial information about the electronic properties of the material, such as the presence of a band gap or the nature of the conduction and valence bands. Half-Heusler compounds often exhibit complex band structures due to the interaction between the different atomic orbitals. The position and width of the band gap, the effective mass of charge carriers and the presence of multiple conduction bands all play significant roles in determining the material's thermoelectric performance. By analyzing the band structure, researchers can identify optimal compositions and structures that enhance the material's electrical conductivity while minimizing its thermal conductivity [3].
The transport properties of a material, including electrical and thermal conductivity, are crucial for assessing its thermoelectric performance. Electrical conductivity is directly related to the material's band structure and the mobility of charge carriers. High electrical conductivity is desirable for efficient thermoelectric performance, as it enables effective conversion of thermal energy into electrical energy. Thermal conductivity, on the other hand, is influenced by both lattice vibrations (phonons) and electron-phonon interactions. In thermoelectric materials, it is essential to minimize thermal conductivity to improve the thermoelectric figure of merit (ZT). This can be achieved through various strategies, such as alloying, nano-structuring, or introducing point defects to scatter phonons. First-principles calculations can predict these transport properties by combining information from the band structure with models of electron and phonon transport. The Boltzmann transport equation, solved within the framework of DFT, is often used to estimate the electrical and thermal conductivities. This approach allows for the prediction of the Seebeck coefficient, electrical resistivity and thermal conductivity, which are combined to determine the material's ZT value [4].
To illustrate the practical application of first-principles analysis, we consider several case studies of high-performance Half-Heusler thermoelectric materials. These case studies highlight the effectiveness of first-principles methods in guiding the design and optimization of these materials. One example is the study of the Half-Heusler compound NiTiSn, which has shown promising thermoelectric performance due to its suitable band structure and low thermal conductivity. Through first-principles calculations, researchers have been able to identify compositional changes that enhance the material's thermoelectric efficiency. Another example is the exploration of Half-Heusler alloys with dopants or substitutions that modify the electronic structure and improve performance [5].
The first-principles analysis of band structures and transport properties in high-performance Half-Heusler thermoelectric materials provides invaluable insights into the design and optimization of these compounds. By leveraging advanced computational methods and theoretical frameworks, researchers can predict and enhance the thermoelectric performance of Half-Heusler alloys. Through a detailed understanding of the band structure, it is possible to identify materials with favorable electronic properties, such as narrow band gaps or optimal carrier effective masses. This knowledge, combined with insights into transport properties such as electrical and thermal conductivities, allows for the development of materials with high thermoelectric efficiency. The application of first-principles calculations has already led to significant advancements in the field of thermoelectrics, with numerous case studies demonstrating the potential for improved performance. As computational methods continue to evolve and refine, the ability to predict and design highperformance thermoelectric materials will only increase, opening new avenues for waste heat recovery and solid-state cooling technologies. In conclusion, first-principles analysis serves as a powerful tool in the study of Half-Heusler thermoelectric materials, offering a deeper understanding of their electronic and transport properties. This approach not only aids in the identification of promising materials but also guides the optimization of their performance, paving the way for future advancements in thermoelectric technology.
None.
None.
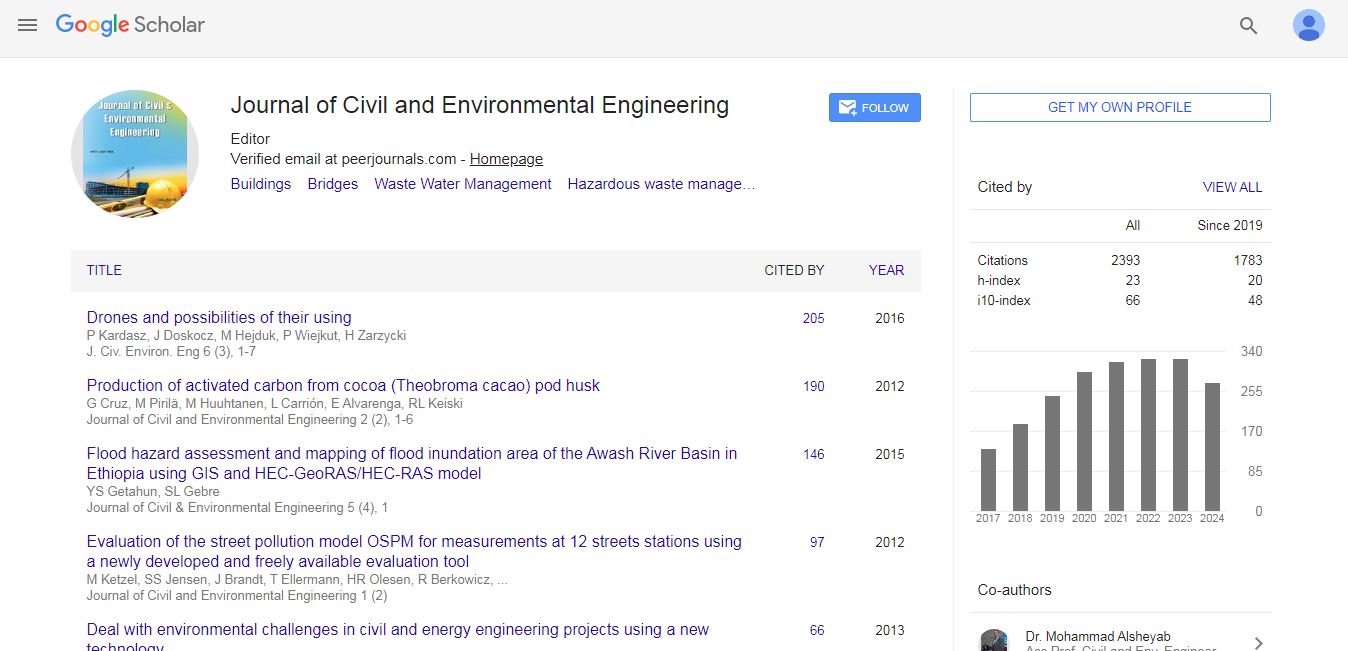
Journal of Civil and Environmental Engineering received 1798 citations as per Google Scholar report