Opinion - (2022) Volume 8, Issue 4
Received: 02-Jul-2022, Manuscript No. jotr-22-76293;
Editor assigned: 04-Jul-2022, Pre QC No. P-76293;
Reviewed: 18-Jul-2022, QC No. Q-76293;
Revised: 23-Jul-2022, Manuscript No. R-76293;
Published:
28-Jul-2022
, DOI: 10.37421/2476-2261.2022.8.207
Citation: Toyokuni , Zukile. “Genetic Instability and Carcinogenesis are promoted by Caspase-3.” J Oncol Transl Res 8 (2022): 207.
Copyright: © 2022 Toyokuni Z. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Apoptosis, or modified cell passing, is the most obvious method of cell demise in multicellular creatures. A significant physiological capability of apoptosis is to dispose of harmed or undesirable cells in early turn of events or to keep up with physical tissue homeostasis at later stages. Accordingly, it is for the most part expected that apoptosis is an enemy of cancer-causing process because of its fundamental job in eliminating cells that have endured DNA damag. DNA harm and resulting changes in key oncogenes and growth silencer qualities is a key cycle prompting disease. Thusly, the ongoing worldview is that apoptosis is a boundary for carcinogenesis. For instance, numerous growth silencer qualities changed in disease frequently have apoptosis-advancing capabilities. Instances of these incorporate p53, PTEN, BAX, and INK4a/ARF. Changes in these qualities frequently permit harmed cells to endure when they ought to pass on. Then again, numerous oncogenes whose articulations are many times improved in malignant growth cells have hostile to apoptotic capabilities. Instances of these incorporate Bcl2, Bcl-xL, Akt/PKB and mTOR [1].
In any case, there is expanding acknowledgment that the contribution of apoptosis in carcinogenesis might be more muddled. A few oncogenes seem to sharpen cells to apoptosis. For instance, the apoptosis-advancing elements of c-Myc and E1A are irrefutably factual in various examinations. In any case, the job of apoptosis in carcinogenesis actuated by these qualities isn't surely known. For both c-Myc and E1A, their favorable to apoptotic exercises seem coupled to their hyperproliferative exercises. In this manner, it was proposed that apoptosis goes about as a safeguard component to restrict the results of unusual mitogenic flagging. There was likewise an idea that oncogeneinterceded unreasonable apoptosis could make a determination strain to beat apoptosis, in this manner permitting disease cells to turn out to be more dangerous. In any case, these theories have not been tentatively assessed [2].
Proof is arising that a few variables in the apoptotic hardware might assume facilitative parts in carcinogenesis. A model is Fas Ligand (CD95), which is a key part in intervening the outward pathway of cell apoptosis. Hereditary knockout of Fas Ligand weakened tumorigenesis in mice as opposed to advancing it. Fas Ligand was displayed to advance carcinogenesis by actuating downstream c-Jun and JNK pathway. Fas Ligand has likewise been displayed to advance malignant growth cell intrusiveness and metastasis by associating and actuating c-Met oncogene. Another new review showed that more elevated levels of enacted caspase-3 in head and neck disease or bosom malignant growth were connected with expanded post treatment cancer repeat or patient passing, in spite of the customary way of thinking [3].
In this review, we planned trials to analyze the irrational speculation that caspase-3 works with carcinogenesis by prompting hereditary unsteadiness in cells that endure cytotoxic pressure. We gave solid proof that enacted caspase-3 could for sure advance oncogenic change in human cells and in mice by prompting tireless hereditary shakiness through its downstream effector, endonuclease G, whose ordinary capability is to piece genomic DNA in apoptotic cells. Caspases are urgent chemicals in apoptosis through which harmed or undesirable cells deliberately annihilate their own foundation to end it all. Enactment of caspases in cells presented to cytotoxic pressure (e.g., radiation) is generally viewed as an irreversible cycle that will bring about the end of host cells. Subsequently, it has been by and large expected that all cells with apoptotic caspase enactment will pass on. Notwithstanding, information in this regard are missing for mammalian cells. We chose to complete a nitty gritty examination of the connection between caspase-3 enactment and the destiny of host cells, since caspase-3 is involved toward the end phase of apoptosis. To do this, we utilized a painless caspase-3 correspondent as of late evolved in our lab. In this correspondent, the EGFP quality is combined with a firefly luciferase quality journalist through an adaptable linker. The combination protein is additionally connected up with a polyubiquitin space, which delivers the combination correspondent protein entirely unsteady since it is perceived by the proteasome framework as a ubiquitylated protein and quickly corrupted. In the middle between the EGFP-luc and polyubiquitin spaces, a caspase-3 cleavage site is embedded. Under typical conditions consistent state correspondent protein levels will be extremely low in have cells. Nonetheless, when caspase-3 is initiated, the polyubiquitin area will be cut off and the EGFPLuc combination protein will be settled [4, 5].
To additionally portray the destiny of individual cells with caspase-3 initiation, MCF10A cells transduced with caspase-3 journalists were lighted with various dosages of radiation and broke down through stream cytometry. The cells were then figured out eight distinct reaches as indicated by caspase-3 correspondent enactment levels. Higher radiation dosages expanded the small amount of cells with more significant levels of caspase-3 initiation. Cells from various reaches were then arranged one cell/well into various wells of 96- well plates. EGFP articulation in the wells was additionally affirmed however fluorescence microscopy (information not shown). Every one of the singular wells inspected showed positive recognizable proof of one cell/well. Likewise, EGFP articulation was seen obviously in cells from M4-M8 ranges. Province development from the exclusively plated cells with various GFP articulation status (M1-M8) was then dissected following 2 weeks in the 96-well plates. True to form, there was a reasonable radiation portion subordinate lessening in settlement framing capacities. Notwithstanding, the connection between caspase-3 actuation and state framing capacities was more confounded. It gave the idea that cells could endure an extensive variety of caspase-3 enactment levels particularly at lower dosages of radiation. At a moderate radiation portion (3Gy), the connection between caspase-3 initiation and clonogenic endurance become direct, with M4 as the edge. At more elevated levels (>3Gy), a large portion of the cells couldn't shape states regardless of caspase-3 journalist exercises, reliable with the capacity of radiation to kill cells in an apoptosis-free way.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
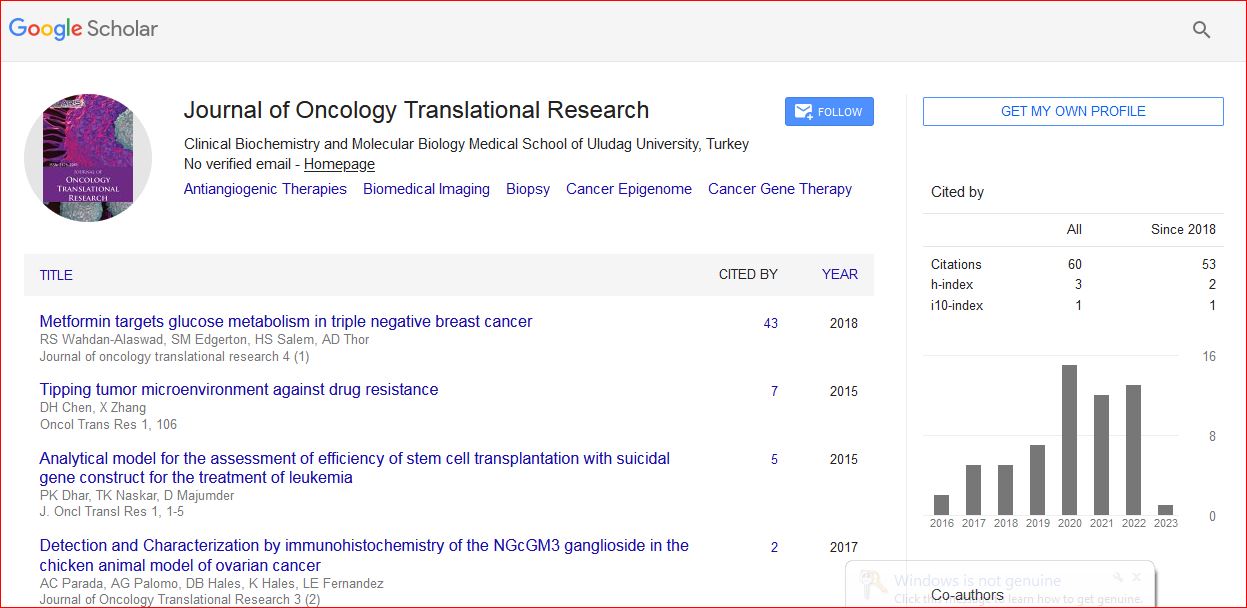
Journal of Oncology Translational Research received 93 citations as per Google Scholar report