Opinion - (2022) Volume 11, Issue 12
Received: 25-Nov-2022, Manuscript No. jhoa-23-86319;
Editor assigned: 28-Nov-2022, Pre QC No. P-86319;
Reviewed: 13-Dec-2022, QC No. Q-86319;
Revised: 19-Dec-2022, Manuscript No. R-86319;
Published:
27-Dec-2022
, DOI: 10.37421/2167-1095.2022.11.380
Citation: Sounira, Mehri. “Hypertension in the Lungs in Sickle Cell Disease Diagnostics and Treatment.” J Hypertens 11 (2022):380.
Copyright: © 2022 Sounira M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Pulmonary hypertension affects approximately 10% of adult sickle cell disease (SCD) patients, particularly those with the homozygous genotype. A rise in pulmonary artery systolic pressure, which can be measured noninvasively by echocardiography, helps identify SCD patients at risk for pulmonary hypertension, even though right-heart catheterization is required for a definitive diagnosis. About half of patients with SCD-related pulmonary hypertension have precapillary pulmonary hypertension, which could be caused by either (1) a nitric oxide deficiency state and vasculopathy as a result of intravascular hemolysis, (2) chronic pulmonary thromboembolism, or (3) upregulated hypoxic responses as a result of anemia, low O2 saturation, or microvascular obstruction. Left ventricular dysfunction causes postcapillary pulmonary hypertension in the remaining individuals. Even though their pulmonary artery pressure is only slightly elevated, SCD patients with pulmonary hypertension have a significantly higher risk of death than SCD patients without pulmonary hypertension. Guidelines for the diagnosis and treatment of pulmonary hypertension caused by SCD were recently published by the American Thoracic Society [1]. Anticoagulation is used to treat thromboembolism in adults with sickle-related pulmonary hypertension; oxygen therapy is used to treat low oxygen saturation; postcapillary pulmonary hypertension patients are treated for left ventricular failure; and hydroxyurea or transfusions are used to increase haemoglobin concentration, reduce hemolysis, and stop vaso-occlusive events that increase pulmonary pressure. Randomized trials have not identified any medications that can lower pulmonary pressure in SCD patients with precapillary pulmonary hypertension. Treatments that have been shown to be effective in treating other types of pulmonary arterial hypertension should be considered for patients with hemodynamic pulmonary arterial hypertension [2]. It is said that some of these treatments can lower pulmonary hypertension caused by SCD. Compound heterozygosity, such as haemoglobin SCD and haemoglobin S-thalassemia, or homozygosity for the Glu6Val mutation in HBB (sickle cell anaemia; SCD) can cause sickle cell disease (SCD). SS hemoglobin). Haemoglobin S, the result of this mutation, is a structurally abnormal form of hemoglobin. Haemoglobin S polymerization is the primary factor that contributes to the negative effects of hemolysis, vaso-occlusion, chronic inflammation, anemia, and an increase in hypoxic responses. It is essential to take into consideration both the prevalence of pulmonary hypertension in patients with SCD and the broader picture of pulmonary hypertension in patients without SCD because pulmonary hypertension influences the pathogenesis, classification, and prognosis of SCD.
The World Symposium on Pulmonary Hypertension defines pulmonary hypertension as a right cardiac catheterization-measured mean pulmonary artery pressure of 25 mm Hg at rest. People whose mean pulmonary artery pressure is between 21 and 24 mm Hg are being questioned regarding their therapeutic significance. Mean pulmonary artery pressure can never go above 20 mm Hg, which is considered normal. People who fall into this range have a lower exercise capacity and are less successful. This range is regarded as a description of borderline pulmonary hypertension by some authorities. There are five main clinical categories for pulmonary hypertension [1]. In Group 1 of its 2009 guidelines, the European Society of Cardiology and the European Respiratory Society Task Force for the Diagnosis and Treatment of Pulmonary Hypertension include pulmonary arterial hypertension associated with SCD and other haemolytic anemias. During the Fifth World Symposium on Pulmonary Hypertension in 2013, the decision was made to move pulmonary hypertension with multiple or unknown etiologies, including SCD, from Group 1 to Group 5. The pathological intima, medium, and adventitia of the pulmonary arteriolar wall are affected by pneumatic arterial hypertension in Group 1. A rise in the number and size of smooth muscle cells in the media surrounding the vessel wall results in medial hypertrophy. The movement of smooth muscle cells from the media to the endothelial layer that typically lines the artery lumen is what causes intimal proliferation. Plexiform lesions in the artery lumen are caused by the growth of endothelial cells and a layer of myogenic cells in the interstitial space. The expansion of cells surrounding the media, such as fibroblasts, progenitor cells, macrophages, and other immune cells, thickens the adventitia, which serves as a resource for the healing of vessel damage [2]. Changes similar to those seen in pulmonary arterial hypertension can also occur in other forms of pulmonary hypertension, albeit to varying degrees. In Group 2 pulmonary hypertension caused by left heart dysfunction, the pulmonary veins and capillaries are also swollen. In Group 4 chronic thromboembolic pulmonary hypertension (CTEPH), organized thrombi adhere to the medial layer, replace the intima of the proximal or distal elastic pulmonary arteries, and either partially or completely block the lumen. Systemic circulation collateral arteries may expand to completely occluded locations, and changes like pulmonary arterial hypertension may not manifest in occluded areas. Depending on whether the right heart catheterization measured the pulmonary capillary wedge pressure (PCWP) or the left ventricular end-diastolic pressure (LVEDP) at a value that was less than or equal to 15 mm Hg, pulmonary hypertension is further divided into precapillary pulmonary hypertension and postcapillary pulmonary hypertension when it comes to hemodynamics [3]. The clinical classification of Group 2 pulmonary hypertension and the hemodynamic classification of postcapillary pulmonary hypertension both refer to the same condition in practice.
The tricuspid regurgitation velocity (TRV) measured during echocardiography in conjunction with the predicted right atrial pressure is generally considered to be a reliable estimate of systolic pulmonary artery pressure. Pulmonary hypertension is defined as a mean pulmonary arterial pressure of 2.5 m/sec, while the average pulmonary arterial pressure is thought to be 25 mm Hg. However, estimates of systolic pulmonary artery pressure and TRV elevation do not accurately identify individuals with pulmonary hypertension [4]. Instead, patients who require a right cardiac catheterization for a more in-depth examination can be identified with an elevated TRV. Pulmonary hypertension is regarded as unlikely if the TRV is less than 2.8 m/sec and there are no additional echocardiographic changes that could indicate it, such as right-sided chamber expansion or right ventricular systolic dysfunction. If the TRV is greater than 3.4 m/sec, the diagnosis is considered likely, and if it is between 2.9 and 3.4 m/sec, it is considered plausible.
As determined by TRV during Doppler echocardiography, approximately 30% of individuals with haemoglobin SS and 10% to 25% of adults with haemoglobin SC had elevated systolic pulmonary artery pressure. In addition, more than half of people with haemoglobin SS who do not have a higher TRV at rest experience unusually high PA systolic pressure during activity. When systolic pulmonary pressure rises even slightly, adults with haemoglobin SS have lower exercise tolerance and lower survival rates. Additionally, systolic pulmonary artery pressure is elevated in 10% to 15% of children with SCD; This observation appears to be associated with a decreased capacity for activity, although the implications for survival are unknown. Adults with elevated systolic pulmonary artery pressure at right heart catheterization who do not have pulmonary hypertension have similar survival rates to SCD patients with elevated TRV, indicating that the subset of patients who do have pulmonary hypertension is primarily responsible for the elevated mortality associated with high TRV. A striking clinical feature of sickle-related pulmonary hypertension is its high mortality, despite having relatively low mean pulmonary artery pressures. Patients with SCD have mean pulmonary pressures in the range of 30 to 40 mm Hg, with mild elevations in pulmonary vascular resistance, in contrast to other patients with pulmonary arterial hypertension (such as idiopathic and scleroderma-associated patients), in which morbidity and mortality are typically associated with mean pulmonary arterial pressures in the range of 50 to 60 mm Hg [5-7].
Non-SCD patients with pulmonary arterial hypertension have a negative mortality correlation between their cardiac output and the condition. SCD patients with or without pulmonary hypertension have significantly higher cardiac output than nonanemic individuals. This theory suggests that people with SCD-related pulmonary hypertension might live longer than people without SCD who have pulmonary arterial hypertension, but this is not the case. Patients with anemia who are close to the limits of cardiac output compensation experience significant morbidity and mortality from any level of pulmonary hypertension. As a result of recurrent SCD effects like pain crises81 and, particularly, acute chest syndrome, SCD patients with pulmonary hypertension also experience "acute on chronic" pulmonary hypertension, which reduces their likelihood of surviving these sickle-related issues. "There is insufficient evidence to make a recommendation supporting regular screening with Doppler echocardiography for pulmonary hypertension because studies demonstrating benefit of treating pulmonary hypertension are not available," the National Heart, Lung, and Blood Institute's Expert Panel stated in 2014. In contrast, the Ad Hoc Committee on Pulmonary Hypertension and Sickle Cell Disease of the American Thoracic Society stated in the same year that "committee members routinely perform risk stratification on their patients with SCD by measuring the TRV via Doppler echocardiography" and suggested screening every one to three years. The objective of the American Thoracic Society's Ad Hoc Committee was to establish standard procedures for treating patients with pulmonary hypertension who had high-risk SCD. Here, we summarize the Ad Hoc Committee's suggested guidelines for assessing SCD patients for pulmonary hypertension. In non-SCD pulmonary arterial hypertension patients, even mild anemia has a negative impact on survival. Despite increasing cardiac output, a hemodynamic feature that predicts longer survival, anemia was associated with a higher mortality rate. Even though, as was mentioned earlier, the mean pulmonary artery pressure is only slightly elevated; The high mortality rate of SCD patients with pulmonary hypertension may be exacerbated by anemia in SCD patients. Case reports indicate that SCD patients with pulmonary hypertension benefit from exchange blood transfusion, an intervention that reduces anemia.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
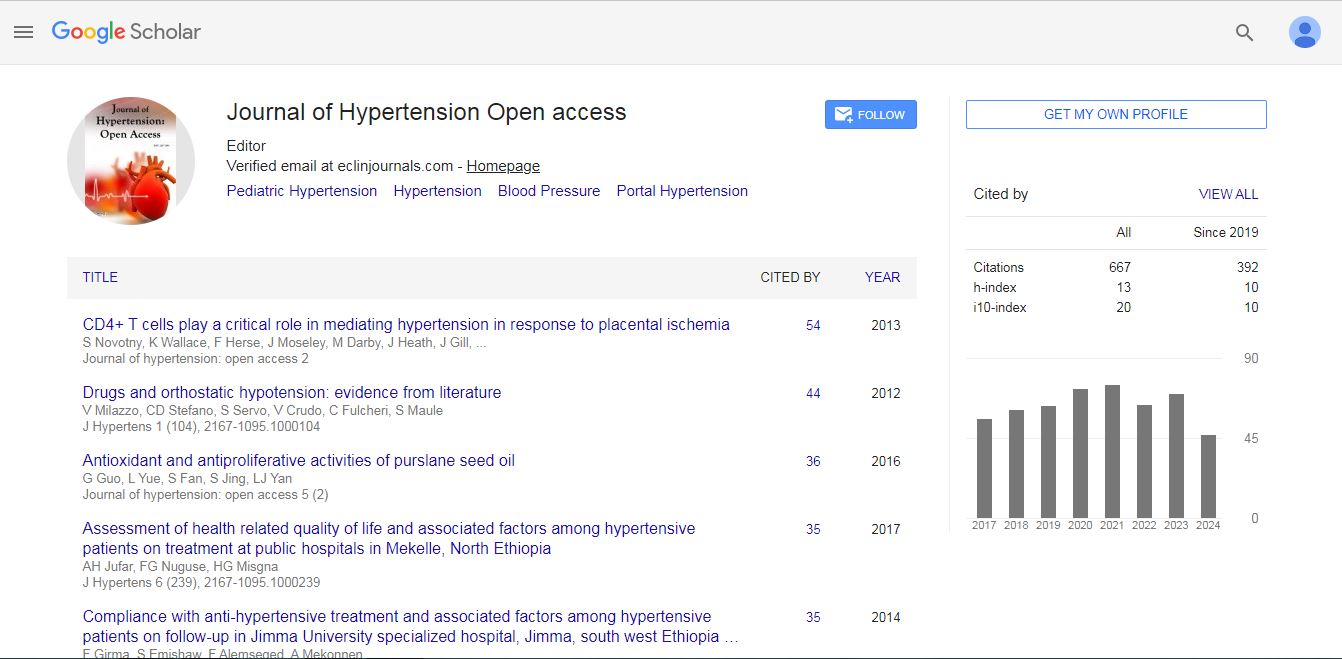
Journal of Hypertension: Open Access received 614 citations as per Google Scholar report