Method - (2023) Volume 9, Issue 2
Received: 01-Mar-2023, Manuscript No. JEFC-23-91296;
Editor assigned: 03-Mar-2023, Pre QC No. P-91296;
Reviewed: 15-Mar-2023, QC No. Q-91296;
Revised: 20-Mar-2023, Manuscript No. R-91296;
Published:
27-Mar-2023
, DOI: 10.37421/2472-0542.2023.9.437
Citation: Lencho, Gemechu Warkina. “Laboratory Manual for Food Analysis." J Exp Food Chem 9 (2023): 437.
Copyright: © 2023 Lencho GW. This is an open-access article distributed under the terms of the creative commons attribution license which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
This manual is an updated version of laboratory manual for Food Analysis. It has been compiled to fill in the gap in the existing old manual used in the laboratory of the Food Science and Postharvest Technology Department. The old version was a fragmented piece of protocols for each parameter and creates difficulty for a reader who needs to understand the method principle of each parameter to be measured in detail. The scope of this manual has taken into account the details of proximate analysis methods (moisture content, crude protein, crude fat, crude fiber, and ash content and total carbohydrate); bioactive determination methods (polyphenol, total flavonoid, ascorbic acid, and beta-carotene) and method of total antioxidant capacity determination. Furthermore, this updated version of the manual covers the basic principles and analytical procedures applied in the analysis of proximate, bioactive components and antioxidant capacity as applied to food biomaterials. A great effort has been made by the contributor of this document to make it very easy so as to make it a user friendly type manual for the readers and users.
Food analysis • Analytical method • Proximate • Bioactive component • Total antioxidant
Proximate analysis
Determination of moisture content
Principle: When heat is provided to the food sample by electrically heated air oven, the moisture evaporates which leads to a decrease the sample’s weight since the weight of water is evaporated. This decrease in sample weight can help us to calculate the difference in the weight between the original sample and the oven-dried sample. Then, the percent of moisture is determined from weight difference between initial and final mass of food sample [1].
Apparatus:
• Dishes made of aluminum or stainless steel approximate 120 mm of diameter and 2.5 mm deep with tight fitting lids
• Hot air oven
• Analytical balance
• Desiccator
• Spatula
Procedure:
• Dry clean glass petridish in oven at 105°C for 20 minutes.
• Take out the petridish from oven and cool it in desiccator. Next, weigh the empty dishes and lid.
• Homogenize the flour sample thoroughly and transfer about 5 g to petridish.
• Remove the lid; place the dish and the lid in the oven at 135°C for 2 hour and avoiding contact of the dish with walls.
• Remove the dish from the oven, replace the lid, cool in desiccator and reweigh when cooled.
• Dry further to make sure that a constant weight has been achieved.
• Finally, determine the moisture content of the sample using the following calculation:
Calculation:
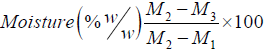
Where, M1= mass of empty dish
M2=mass of empty dish plus mass of sample before drying
M3=mass of empty dish plus mass of sample after drying
Protein content determination
Principle: The food sample to be analyzed is weighed into a digestion flask and digested by heating it in the presence of concentrated sulfuric acid as oxidizing agent, anhydrous potassium sulfate (to speed up the reaction by raising the boiling point) and a catalyst, such as copper (speed up the reaction). Digestion converts all nitrogen in the food into ammonia, and other organic matter to CO2 and H2O. Next, the solution in the digestion flask is made alkaline by addition of sodium hydroxide which is steam distilled and converts the ammonium sulfate into ammonia gas. Ammonia gas is liberated from the solution and moves out of the digestion flask to the receiving flask which contains an excess of boric acid. Then, the low pH of the solution in the receiving flask converts the ammonia gas into the ammonium ion and simultaneously converts the boric acid to the borate ion [2]. Finally, the nitrogen content is estimated by titration of the ammonium borate with standard hydrochloric acid using a suitable indicator to determine the end-point of the reaction.
Chemicals:
• CuSO4 (cupper (II) sulfate)
• K2SO4 (potassium sulfate)
• 98% H2SO4 (sulfuric acid)- specific gravity=1.84 g/mL
• 4% boric acid
• 40% NaOH (sodium hydroxide)
• 0.2 N HCl (0.2 normality of hydrochloric acid)
• Bromocresol green and methyl red
Apparatus and equipment:
• Kjeldahl digester- DK 6 or DK 12
• Kjeldahl digestion flask – 500 mL
• Distillation and titration apparatus- (VELP SCIENTIFA nitrogen analyzer, Italy)
• Burette- 50 mL
Preparation of chemical standards:
• 40% NaOH- dissolve 40 g of NaOH in 100 mL of distilled water and mixed it gently
• 4% boric acid: dissolve 4 g of boric acid in 100 mL of distilled water
• Brocressol green indicator – dissolve 0.1 g of bromocressol green powder in 100 mL of ethanol
• Methyl red indicator – dissolve 0.1 g of methyl red powder in 100 mL ethanol
• Hydrochloric acid (0.2N HCl):- add 16.6 mL of 37% HCl to 1000 mL of distilled water in flask.
To determine the molarity of a mass percent solution, use the following procedure:
• Determine the mass of solution by multiplying the volume of the solution by the density of the solution.
Mass of solution=volume of solution × density of solution
• Determine concentration in percent by mass of the solute in solution. Change to the decimal equivalent.
• Calculate the molar mass of the compound
Multiply mass (step 1) by mass% (step 2) and divide by molecular mass (step 3) to find the number of moles present in the whole solution.
• Divide the number of moles (step 4) by the volume in liters of the solution to find the molarity of the solution.
Determine molarity of 37% hydrochloric acid (specific density 1.19 g/mL):
• Mass of solution=1,000 mL × 1.19 g/mL=1,190 g
• Mass%=37%=0.37
• Molar mass of hydrochloric acid=36.5 g/mol
• Mass × mass%=1,190 g × 0.37=440.3 g
• Moles=given mass/molar mass=440.3 g/36.5 g/mol=12.06 moles
• Molarity=moles/liters=12.06 moles/1 liter=12.06 M (M=N, since HCl is monoprotic acid)
Normality of 37% HCl=12.06N
N1V1=N2V2 (dilution calculation)
12.06 V1=0.2* 1000ml (1L=1000 mL)
V1=16.6 mL
Procedure:
a) Weigh 1 g (5 mL) of food sample by analytical balance and add 0.2 g of CuSO4 and 7 g of K2SO4 catalysts to each sample.
b) Add 15-25 mL 98% concentrated H2SO4 to each sample. Put the test tube containing sample in heating block of digester at 420°C for 1:30 hour until all color removed (disappearance of color).
c) Then, Switch off digester and wait until it cools. Neutralize the digested sample by using 40% NaOH and titrate it by 0.2N HCl.
d) Finally, calculate percent of protein from total nitrogen by calculation.
Calculation:
Total nitrogen(%) = * (V −Vb)* N*14 +W ×100
Where,
V= volume of acid consumed to neutralized the sample
Vb= the volume of acid consumed to neutralize the blank
N=normality of the acid
14=Equivalent weight of nitrogen.
Crude protein (%)=(total nitro (%) * conversion factor)
In case of dairy ice cream/kulfi, calculate milk protein as N% × 6.38
In case of frozen dessert and most food samples; calculate total protein as N% × 6.25
In case of high protein foods, calculate total protein as N% × 5.51
Determination of ash content
Principle: Principle involved is that when a known weight of food or feed is ignited at high temperature (550°C) and complete oxidation of organic matter, then the inorganic residue remaining after complete oxidation expressed ash content [3].
Apparatus and equipment:
• Analytical Balance
• Crucible
• Muffle furnace
• Safety tongs
• Desiccators
Procedure:
a) Dry clean crucible in oven at 105°C for 20 minutes.
b) Take out the crucible from oven and cool it in desiccator. Next, take note the Weight of tarred crucible and transfer about 3 g of sample to crucible.
c) Place crucibles in cool muffle furnace and burn for 12-18 hours (or overnight) at 550°C.
d) Turn off muffle furnace and allow cooling to at least 250°C. Open door gently to avoid losing ash that may be fluffy.
e) Transfer crucibles to desiccator by using safety tongs and cover crucibles and close desiccator and cool to room temperature and then reweigh each crucibles containing ash.
f) By difference, calculate the ash content of the sample by the following formula and expressed in percentage.
Calculation:
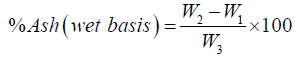
Where, W1= weight of empty crucible
W2= weight of crucible containing ash
W3= sample weight before ashing
Crude fat determination
Principle: Crude fat content is determined by extracting the fat from the sample using either petroleum ether or normal hexane as a solvent from dried sample in a Soxhlet apparatus, and the other is evaporated from the extraction flask. The crude fat content is determined from the difference in weight of the extraction flask before and after extraction [4].
Chemicals:
• Petroleum spirit boiling point 60-80°C
• Acid washed sand
Apparatus and equipment:
• Analytical balance (at least 1 mg sensitivity)
• Electrical drying oven to be operated at 102°C ± 1°C
• Soxhlet extraction unit comprising: - Round bottom flask, 150 mL, Soxhlet extractor with 60 mL siphoning capacity and condenser
• Cellulose extraction thimbles (28 × 80 mm)
• Fume cupboard
• Heat source, either electric heating mantle, or steam bath 100 mL beaker
• Desiccator with silica gel desiccant
• Glass rod
• Cotton wool free of fat
Procedure:
a) Rinse all glassware with petroleum spirit, drain, dry in an oven at 102°C or 30 min and cool in a desiccator.
b) Put a plug of cotton wool in the bottom of an extraction thimble to avoid the escape of material from thimble and stand the thimble in the beaker.
c) Accurately weigh 5 g of sample into the thimble and add 1-1.5 gram of sand and mix the sand and sample with a glass rod.
d) Wipe the glass rod with a piece of cotton wool and place cotton wool in the top of the thimble. (Addition of sand is not required for analysis of meat meal).
e) Dry the sample in an oven at 102°C for 5 hours. Take the piece of cotton wool from the bottom of the beaker and place it in the 3 top of the thimble and insert the thimble in a Soxhlet liquid/solid extractor.
f) Accurately weigh a clean, dry 150 mL round bottom flash and put about 90 mL of petroleum spirit into the flask.
g) Assemble the extraction unit over either an electric heating mantle or a water bath and heat the solvent in the flask until it boils after adjust the heat source so that solvent drips from the condenser into the sample chamber at the rate of about 6 drops per second and continue the extraction for 6 hours. For sausage meat and other emulsified products, the extraction should be performed in stages: Extract for about 4 hours, then remove the heat source and drain the solvent from the extractor in the flask.
h) Remove the thimble from the extractor and transfer the sample to a 100 mL beaker.
i) Break up the sample with a glass rod and return the sample to the thimble and replace the thimble in the extractor.
j) Rinse the beaker with petroleum spirit and pour rinsing into the extract for a further two hours. Remove the extraction unit from the heat source and detach the extractor and condenser and replace the flask on the heat source and evaporate off the solvent (the solvent may be distilled and recovered).
k) Place the flask in an oven at 102°C and dry the contents until a constant weight is reached (1-2 hours).
l) Finally, cool the flask in a desiccator and weigh the flask and calculate crude fat of the sample.
Calculation:
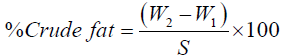
Where, Weight of empty flask (g)=W1
Weight of flask and extracted fat (g)=W2, Weight of sample=S
Determination of crude fiber
Principle: For crude fiber determination, the food sample should be ground finely or crushes for easy interaction with chemicals and extract the fat from food before estimating the crude fiber to facilitate the process of digestion and filtration. Then, crude fiber is determined gravimetrically after chemical digestion and solubilization of other materials present by treating the sample with a boiling sulfuric acid and subsequently with boiling potassium hydroxide or sodium hydroxide. The residue remaining after ignition of the residue and subtraction of ash is referred to as fiber [5].
Chemicals:
• 28% potassium hydroxide
• 1.25% H2SO4 (sulfuric acid)
• 1.25% NaOH (sodium hydroxide)
• Acetone
Apparatus and equipment:
• Beaker- 600 mL
• Buchner funnel fitted with a No.9 rubber stopper.
• Conical flask- 250 mL
• Crucible
• Muffle furnace
• Drying oven
• Desiccator
Procedure:
a) Weigh 0.6 g of the sample in each of 600 mL beaker and 200 mL of 1.25% sulfuric acid solution to each beaker and allow boiling for 30 min by rotating and stirring periodically.
b) After 30 minutes, add 20 mL of 28% potassium hydroxide solution in to each beaker and allow boiling for another 30 min. keep the level by addition of hot distilled water. Then after 30 min, filter the solution found in each of the beaker through crucibles containing sand by placing each of them on buchner funnel fitted with no.9 rubber stopper.
c) Wash the sample with distilled water during filtration and wash the final residue with 1.25% sulfuric acid solution, hot distilled water, 1.25% sodium hydroxide solution and finally with acetone.
d) Dry the crucibles with their content in oven for two hour at 130°C and cool it in desiccators and weigh as (W1).
e) Burn or ignite the crucibles with their content in muffle furnace for 30 minutes at 550°C and cool it in desiccators.
f) Finally, weigh the mass of each crucible and take note as (W2). Then, calculate the crude fiber of the sample.
Calculation:
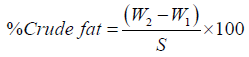
Where, W1=Weight of (crucible+sample) after drying
W2=weight of (crucible+sample) after ashing
W3=Weight of fresh sample
Determination of total carbohydrate content
The carbohydrate content of the sample was calculated by difference rather than direct analysis [4].
Carbohydrate (%) = 100-(Moisture+Ash+Protein+Fat+Fiber).
Analysis of bioactive components and antioxidant capacity
Determination polyphenol content
Principle: The principle of folin- ciocaltea assay is the reduction of the folin–ciocalteu reagent (FCR) which formed from a mixture of phosphor tungstic acid and phosphor molybdic acid after oxidation of the phenols is reduced to a mixture of blue oxides of tungsten and molybdenum. The phenolics resulting in the production of molybdenum–tungsten blue is quantified spectrophotometrically at 760 nm and the intensity increases linearly with the concentration of phenolics in the reaction medium [6].
Chemicals:
• 2N folin-Ciocalteu
• 7.5% sodium carbonate (Na2CO3)
• 99.8% methanol
• 1 mg/mL gallic acid
Apparatus and equipment:
• Centrifuge (2-16 KC)
• UV/VIS spectrometer - (T80 UV/VIS spectrometer, PG instrument Ltd)
• Filter paper
• Mechanical shaker- ((HY-2(A)
Preparation of sample:
• Weigh 0.1 gram of sample and mix with 20 mL of methanol
• Next, shakes for 24 hour on a mechanical shaker
• Then, filter by using filter paper
• Finally, store the extracted samples in a refrigerator (4°C) until analysis
Procedure:
a) Mix 10 mL of the extracted sample with 2 mL of 2N folin-ciocalteau reagent (Prepare by diluting 3 mL of folin reagent ten tmes) and immediately, add 2 mL of 7.5% sodium carbonate solution.
b) Incubate the mixture for 30 minutes at 37°C and read the absorbance by UV-Vis spectrophotometer (T80 UV/VIS spectrometer, PG instrument Ltd) at 760 nm wavelength. Use gallic acid as a standard and the correlation between absorbance and gallic acid concentrations give a calibration standard curve.
c) Draw the calibration curve of gallic acid stock solution by accurately weigh 0.1 g gallic acid into a 100 mL volumetric flask and dissolve in 10 mL methanol and the solution make up to the same solvent.
d) Add 0, 0.625, 1.25, 2.5, 3.75, 5, 6.25, 7.5 and 8.75 mL from the stock solution to flask and then, dilute to give 0. 25, 50, 100, 150, 200 and 250 300 and 350 μg/mL of gallic acid in methanol with the final volume 25 mL for each.
e) Add 1 mL of each sample into test tubes and mix with 2 mL of 2 N folin- ciocalteu reagent and 2 mL of 7.5% sodium carbonate and cover the test tubes with aluminum foil and allow standing for 30 minutes at room temperature.
f) Read the absorbance at 760 nm wavelength using UV-Vis spectrophotometer (T80 UV/VIS spectrometer, PG instrument Ltd) expressed as milligrams of gallic acid equivalent per gram of dry weight (mg GAE/100 g d.w).
Calculation:
Phenolic content = (GAEC*V) / W
Where,
GAEC=concentration of Gallic acid established from calibration curve (mg/mL)
V=Total volume of the extract (mL)
W=weight of extract (g)
Determination of total flavonoid content
Principle: The basic principle of aluminium chloride colorimetric method is that aluminum chloride forms acid stable complexes with the carbon-4 keto group and either the carbon-3 or carbon-5 hydroxyl group of flavones and flavonols. In addition, it also forms acid complexes with the ortho- dihydroxyl groups in the A or B ring of flavonoids. The intensity of the flavonoids-aluminum complex of crude extract was determined by spectrophotometric method at 510 nm by using catechin as a reference standard [7].
Chemicals:
• 1mg/mL D-Catechin
• 1 M NaOH
• 10% AlCl3
• 5% NaNO2
Apparatus and equipment:
• Conical flask-100 mL
• UV/VIS spectrophotometer (T80 UV/VIS spectrometer, PG instrument Ltd)
Sample preparation:
• Dissolve 10 g of sample in 100 mL of 99.8% methanol
• Shake the mixture for 24 hour by multipurpose vibrator
• Centrifuge the mixture at 1000 rpm for 15 minutes and take clear solution through filter paper
• Keep the extracted sample in refrigerator at 4°C until analysis
Procedure:
a) Add an aliquot of each flour sample extract (1 mL) with 0.3 mL of 5% NaNO2 and add
b) 0.3 mL of 10% AlCl3 after 5 min.
c) Add 2 mL of 1 M NaOH and then, add immediately 2.4 mL of distilled water to produce a total volume of 6 mL.
d) Measure the color intensity of the flavonoids-aluminum complex at 510 nm using (T80 UV/VIS spectrometer, PG instrument Ltd) after standing for 10 min at room temperature in dark place.
e) Prepare standard catechin for calibration, weigh about 0.1 g of D-catechin in a 100 mL conical flask and dilute with 10 mL of methanol and make up to the same solvent.
f) Based on the standard curve prepare with catechin equivalent set as 0.0, 6.25, 12.50, 25.00, 50.00, 100.00 μg/mL with final volume 25 mL, calculate the amount of total flavonoid content in extracted samples and express the result as mg of catechin equivalent (CE)/100 g of sample extract on dry matter (DM) basis.
Calculation:
Flavonoid content = (DE*V) / W
Where, DE=D-catechin equivalent (mg/mL)
V=total volume of the sample
W=sample weight (g)
Determination of vitamin C (Ascorbic acid) by titrimetric method
Principle: The ascorbic acid content of a sample can be determined by redox titration method. During this process, the ascorbic acid is oxidized to dehydro-ascorbic acid, which in acidic medium, gives a light pink color. Ascorbic acid reduces oxidation-reduction indicator dye 2, 6 dichlorophenol indophenol to colorless solution [8]. At end point excess unreacted dye is rose pink in acid solution. Extract vitamin and perform titration in presence of metaphosphoric acid -acetic acid solution to maintain proper acidity and avoid auto oxidation of ascorbic acid at high pH.
Chemicals:
• Metha phosphosphoric acid (HPO3)
• Acetic acid (CH3COOH)
• 2,6 dichloro indophenol
• Sodium bicarbonate (NaHCO3)
• Ascorbic acid as standard
Apparatus:
• Conical flask- 200 mL, 500 mL
• Volumetric flask-50 mL
• Burette with stand-100 Ml
Sample preparation:
a) Sample extract is prepared by blending 10 g of sample in the blender and mixed with 50 mL of 5% metaphosphoric acid acetic acid solution and transferred to the 250 mL conical flask.
b) Remaining amount of 50 mL of phosphoric acid solution was added into the flask.
c) Finally, the solution was filtered using Whatman filter paper and the filtrate was collected for determination of vitamin C.
Procedure:
• Preparation of Meta phosphoric acid – acetic acid (HPO3 - CH3COOH) by weighing 15 g of HPO3 sticks and dissolving in 40 mL of CH3COOH and 200 mL of distilled water.
• Dilute the solution to 500 mL with distilled water, filter and store in the dark for use.
• Prepare Indophenols solution by weighting 50 mg of 2, 6-dichlorophenol indophenols powder and dissolve in 50 mL of distilled water containing 42 mg of NaHCO3. Then, dilute the solution to 200 mL with distilled water, filter and store in the dark for use.
• Prepare ascorbic acid standard solution (1 mg/mL) by dissolving 50 mg of ascorbic acid in 40 mL of HPO3 – CH3COOH solution and make up to 50 mL in a volumetric flask. Then, standardize indophenols solution with standard ascorbic acid solution by transferring 2 mL aliquots ascorbic acid solution into 50 mL conical flasks.
• Perform titration of the content of the conical flask with against indophenols from the burette until a distinct rose color persists for about 5 seconds. Blank titration also carried out using 7 mL of HPO3- CH3COOH solution against indophenols.
• Extract Samples by weighing 2 g of each of the powder and fresh samples and transfer into conical flasks.
• Add 40 mL of the extracting solution to each sample and titrate to form a suspension and then, allow standing for 30 minutes. Take note of volume obtain as v mL.
• Filter Sample aliquots (7 mL each) and titrate it with against indophenols from the burette until a distinct pink to rose color persists for 5 seconds. Replicate the titration three times for each sample aliquot and obtain the average titre values.
• Finally, calculate the concentration of ascorbic acid in each sample (mg/g of sample) by using the following formula.
Calculation:
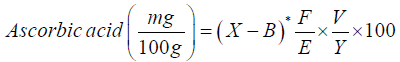
X=Average titre value obtained from sample titration
B=Average titre value obtained from blank titration
F=mg of ascorbic acid equivalent to 1ml of indophenols solution
E=No. of grams of powdered of sample assayed
V=Volume of initial assay solution
Y=Volume of sample aliquot titrated
Determination of beta-carotene content
Principle: Carotenoids are a group of yellow, orange and orange-red fat-soluble pigments and can be extracted into water immiscible solvents like hexane, ethanol and acetone. The absorbance of the extract is measured in a spectrophotometer, and the carotenoids concentration is calculated using a standard curve. β-Carotene is unstable to light and susceptible to airoxidation. Therefore, the sample extracts should be prevented from oxidation and light [9].
Chemicals:
• Beta- carotene standard
• 50% hexane, 25% acetone, 25% ethanol and 0.1% BHT
• CaCl2.2H2O
Apparatus and equipment:
• Conical flask- 50 mL, 100 mL
• UV-Vis spectrophotometer (T80 UV/VIS spectrometer, PG instrument Ltd)
Sample preparation:
• First prepare extraction solvent from 50% hexane, 25% acetone, 25% ethanol, and 0.1% BHT.
• Mix 0.5 g of sample powder with 0.5 g CaCl2.2H2O in analytical conical flask and 50 mL extraction solvent and mix gently for 30 min.
• Add 15 mL of distilled water and the solution frequently shake again for 15 min.
• Separate the organic phases containing the beta-carotene from the water phase, using a separation funnel, and filter by using what man filter paper and use the filtrate sample for analysis.
Procedure:
a) Weigh 0.01 g beta-carotene standard and dissolve in 20 mL solvent which similar to extraction solvent used to extract samples (50% hexane, 25% acetone, and 25% ethanol) and make the volume to 100 mL using the same solvent.
b) Prepare dilution solution from the stock solution (0, 2, 3, 4 and 5 mL) in to 100 mL flask and dilute to give 0, 0.1, 0.2, 0.4, and 0.8 μg/mL of beta- carotene standard in the same solvent.
c) Add 0.5 mL of each extracted sample into test tubes and cover with aluminum foil.
d) Measure the absorbance of the sample extract and betacarotene standard solutions at 450 nm wavelength using UV-Vis spectrophotometer (T80 UV/VIS spectrometer, PG instrument Ltd), then estimate beta-carotene and express in mg/g.
Determination of total antioxidant capacity by DPPH radical scavenging assay
Principle:
DPPH (standard 2, 2-Diphenyl-1-picrylhydrazyl) is Stable free radical and measure the antioxidant activity of tissue extracts. Act as free radical scavengers based on electron-transfer that produces a violet solution in methanol. This free radical is stable at room temperature and reduced in the presence of an antioxidant molecule, giving rise to colorless methanol solution. DPPH reacts with a hydrogen donor of antioxidant and absorb of hydrogen from an antioxidant [10]. Therefore, the antioxidant concentration effect can be easily evaluated by following the decrease of UV absorption at 517 nm.
Chemicals:
• 99% methanol
• DPPH standard (Sigma Aldrich, Germany)
Apparatus and equipment:
• Analytical balance
• Water bath
• Centrifuge (Sigma 2-16KC, UK)
• Micro pipette
• Spectrophotometer (T80 UV/VIS spectrometer, PG instrument Ltd)
Sample preparation:
• Weigh 1g of powdered food sample and add 25 mL 99.8% methanol to the sample and seal the sample by Al foil.
• Put the sample in a shaking water bath (100 rpm, room temperature for 2.5hrs).
• Take the sample after 2.5 hours and centrifuge for 15 min (6000-8000 rpm) and filter the top solution (super natal) part through a filter paper
Procedure:
a) 1M DPPH solution preparation by 4 mg of DPPH with 100 mL of 99.8% methanol and cover the DPPH solution and keep in cool condition.
b) Prepare a solution series using the extracted solution and methanol, take 1 mL from each extracted sample and add 3 mL of DPPH solution to each measured samples and mark up each solution with 99.8% methanol up to 10 mL.
c) Keep the solution in dark place for 30 minute.
d) Measure the absorbance at 517 nm by UV –Visible spectrophotometer (T80 UV/VIS spectrometer, PG instrument Ltd).
e) Calculate the percentage of DPPH free radical scavenging activity by using absorbance difference between sample and control.
Calculation:
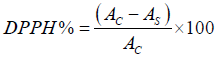
Where; AC=control absorbance (DPPH radical + methanol) or blank solution
AS=sample absorbance (DPPH radical + sample)
W2=weight of filter paper + alkaloid precipitate
Journal of Experimental Food Chemistry received 389 citations as per Google Scholar report