Research Article - (2021) Volume 11, Issue 8
Received: 05-Aug-2021
Published:
26-Aug-2021
, DOI: 10.37421/2161-0444.2021.11.593
Citation: Yuanlin, Liu, Wenda Zhu, Chao Zhang and Yan
bing Li, et al . “N- Acetyl- L-tyrosine enhances the Inhibition Sensitivity of
Procarbazine against Melanoma Targeted Tyrosinase without the Increase
of Toxicity." Med Chem 11(2021): 593.
Copyright: © 2021 Liu Y, et al. This is an open-access article distributed under the
terms of the Creative Commons Attribution License, which permits unrestricted
use, distribution, and reproduction in any medium, provided the original author
and source are credited.
Procarbazine (Pcb) is a component of a chemotherapeutic cocktail in the treatment of melanoma. Unfortunately, Pcb brings about very common adverse effects. In this study, we tried to improve the therapeutically efficacy of Pcb by combination of N-acetyl-L-tyrosine (NAT). We investigated the effect of Pcb/NAT and its underlying mechanism in melanoma via MTT assay, cell cycle assay, ΔΨm assay, Western blotting and in vivo animal model. It was found that combination of Pcb with NAT in 1:1 mole ratio (Pcb/NAT) enhanced the inhibition sensitivity against murine melanoma cells B16/F10 with IC50 value changed from 31.9 ± 1.1 to 14.2 ± 1.1 μM. Similar result was found in K1735 cell line. Compared to Pcb solely, the inhibitory rate of Pcb/NAT against B16/F10-bearing tumor in C57BL/6 mice was 89.4% by 5.8% increase; whilst the volume inhibition rate was 95.6% by 10.1% increase. Pcb/NAT was confirmed with more significant effects on cell differentiation, apoptosis, cycle and mitochondrial membrane potential in B16/F10 cells than Pcb solely. It was found that the present of NAT increased the transformation of Pcb. Involvement of NAT intensified the upregulation of tyrosinase activity/protein level, and the expression of GADPH in vitro and in vivo. Pcb/NAT also enhanced the activation of p53 leading to a more decrease of Bcl-2/Bax ratio than Pcb solely. It was found that sensitivity increase of Pcb/NAT against melanoma brings no increase in toxicity. All the data support that Pcb/NAT is a promising anti-melanoma agent to replace Pcb.
Melanoma • Combination medication • Procarbazine • Tyrosinase • Glyceraldehydes-3-phosphate dehydrogenase
Mutations in melanin-producing nerve sheath cells are the main cause of malignant melanoma, leading to tyrosine metabolism and abnormal melanin production. This type of cancer is a very invasive and malignant solid tumor. The treatment effect of patients in advanced stage is limited and the prognosis is very poor. As a matter of fact, survival time for the median is only 6 to 9 months, and the 1-year survival rate is only 25%. Although the treatment of advanced melanoma has entered the era of targeting and immunotherapy, chemotherapy is still an effective choice [1].
Procarbazine (Pcb) is an alkylating anticancer agent, chemically named as N-isopropyl-α-(2-methylhydrazino)-p-toluamide hydrochloride. Usually, it is administer-ed as a component of a chemotherapeutic cocktail in the treatment of melanoma [2]. Unfortunately, in this combination chemotherapy, Pcb brings about very common adverse effects, especially myelosuppression and reproductive toxicity [2,3]. It is a common sense that the lack of selectivity and sensitivity toward tumor primarily contribute to the toxic side effects of cytotoxic agents. Generally, there are two strategies to reduce side effects. One is to construct the targeted delivery of antitumor agents [4-7]; another is to increase the sensitivity against tumor by combination with certain agents [8-10].
As we know, Pcb contains hydrazine structural moiety, this makes it a kind of prodrugs. In the organism, the hydrazine moiety will be converted to its active form by oxidative enzymes, primarily including cytochrome P450 monooxygenases, and peroxidases [11]. This transformation is the direct oxidation of Pcb. Quite interestingly, there exists an indirect oxidation, which may be realized by the indirect oxidation of mushroom tyrosinase in the presence of phenols or o-diphenols [12]. It was displayed that Pcb was rapidly degraded when submitted to reaction system containing tyrosinase and phenolic substrates (either 4-tert-butylcatechol or N-acetyl-L-tyrosine), where Pcb as well as the phenolic substrate was oxidized [13]. The major oxidation product of Pcb in this process is azoprocarbazine, which is the first active metabolite in previous in vivo study [14]. These evidences imply that either 4-tert-butylcatechol or N-acetyl-L-tyrosine (NAT) may enhance the anti-cancer efficacy of Pcb (Figure S1).
Commonly, NAT is used in place of tyrosine in parenteral nutrition, but human studies indicate considerable amounts of it are excreted unchanged in the urine. It was reported that urinary excretion of NAT in 4 hours accounted for 56% of the infused amount. Nevertheless, there is at least 44% NAT left after 4-hour intravenous infusion. No doubt, NAT is safe in vivo. So, we believe that NAT may possibly work as an anti-cancer promoter with Pcb in vivo based on reasonable administration. Therefore, in the current research, we investigated whether NAT may enhance the antimelanoma efficacy of Pcb in both cellular and animal levels, and what the mechanisms are involved.
Research governance
All animal care and experimental protocols were approved by the Laboratory Animal Ethics Committee of Jinan University (No. 20180226039); All the protocols were also in accordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals (7th edition, USA).
All animal care and experiments were carried on in the Institute of Laboratory Animal Science, Jinan University, China.
Both highly metastatic mouse melanoma B16/F10 and K1735 melanoma cells from mice were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). RPMI 1640 medium was purchased from Life Technologies Ltd. (Grand Island, NY, USA).
PBS buffer was vacuum-filtered through a 0.22 μm membrane before used. Fetal bovine serum (FBS) was obtained from GIBCO (Gaithersburg, MD). Penicillin and streptomycin were from Invitrogen (Carlsbad, CA, USA). Antibodies against Tyrosinase, β-actin, p53, GAPDH, Bcl-2, and Bax were from Cell Signaling Technology (Woburn, USA). All secondary antibodies conjugated with HRP were purchased from Santa Cruz. Methyl thiazolyl tetrazolium (MTT) and all other reagents were from Sigma Chemical (St. Louis, MO). Corning (Corning, NY) offered all culture flasks (Figure S2).
Male C57BL/6 mice were purchased from the Medical Animal Laboratory Centre of Guangdong Province. Mice were 6-8 weeks old and weighed between 18 g and 22 g.
Cell culture
Both B16/F10 and K1735 cells were grown in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS, Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin, and incubated at 37°C in a 5% CO2 humidified atmosphere, respectively. The medium was changed every 1-2 days, and cells were sub-cultured by 0.25% trypsin treatment twice a week. Those cells with passages 4-6 were used in the future experiments.
MTT assay
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, 570 nm) assay was used to measure the inhibition rate of tested samples against B16/F10 and K1735 cellular proliferation. The below process was followed: Cells in suspension at a density of 1×104 cells/well were plated in 96-well plate for 24 h culture, and then were treated for 48 h with addition of either 0.1% DMSO as vehicle or drugs at different concentration. In all experiments, the final DMSO concentration was maintained at a range of less than 0.1% in medium. A concentration range of 0 μm -50 μm (0 μm, 3.125 μm, 6.25 μm, 12.5 μm, 25 μm, and 50 μm) was set for each tested sample and two-fold serial dilutions were applied. Addition of 10 μl MTT solution to each well was then followed, and the plate was incubated for an additional 4 hours. After 150μl of DMSO was added, the absorbance was measured at 570 nm with a microplate reader (Bio-Rad; Hercules, CA, USA). By applying the nonlinear multipurpose curve fitting program GraphPad Prism, the IC50 values for the different treatment conditions were calculated. At least 3 times parallel experiments were pursued on for each test (Figure S3).
Analysis of cell morphology
In a 24-well plate, B16/F10 cells were seeded at 1.5×105 cells/well and incubated with or without Pcb, and Pcb/NAT (in 1:1 mole ratio) at the set concentrations, respectively, and was lasted for 48 h. Afterwards, cell morphology was observed and recorded under the same microscope and lens (Olympus, Japan).
Determination of cell apoptosis and necrosis
In a 6-well plate, B16/F10 cells (2 × 105 cells/ml) were plated, following the addition of either vehicle or the set concentrations of Pcb, and Pcb/NAT (1:1 mole ratio), and cultured for 48 h, respectively. By centrifugation at r. t. and washed twice with ice-cold PBS, the cells were collected. Afterwards, Annexin-V-FITC/PI (KeyGEN; Nanjing, China) was used to stain the cells and flow cytometry (BD FACS Calibur, Franklin Lakes, CA, USA) was applied to analyze cell apoptosis and necrosis.
Cell cycle assay
In a 6-well plate, B16/F10 cells (2×105cells/ml) were seeded, following the addition of either vehicle or the set concentrations of Pcb, and Pcb/NAT (1:1 mole ratio), and cultured for 48 h, respectively. Then, collection of cells was done, following twice washes with PBS. Afterwards, the cells were fixed in cold 70% ethanol (-20 ?) for 12 hours. By centrifugation, the ethanol was removed carefully. Then, suspension of the cells in 1 ml staining reagent (100 mg RNase+50 mg PI/ml) and preservation in darkness for 40 min at r. t. were carried on. By using a flow cytometry (BD FACS Calibur, Franklin Lakes, CA, USA) with an excitation wavelength at 605 nm, cell cycle analysis was then determined.
Mitochondrial membrane potential (ΔΨm) assay
In a 6-well plate, B16/F10 cells (2 × 105 cells/ml) were seeded, following the addition of either vehicle or the set concentrations of Pcb, and Pcb/NAT (1:1 mole ratio), and cultured for 48 hours, respectively. Then, collection of cells was done, following twice washes with PBS. After adding 0.5 ml JC-1 solution and incubating at 37ºC for 30 minutes. The mixture was centrifuged at 800 rpm, washed three times with JC-1 staining buffer. By using flow cytometry (BD FACS Calibur, Franklin Lakes, CA, USA), mitochondrial membrane potential (ΔΨm) of cells was detected (Figure S4).
In a 6-well plate, B16/F10 cells (2 × 105 cells/ml) were seeded and treated with 25 μm of Pcb, and Pcb/NAT (1:1 mol ratio), respectively in a procedure of 24-hours incubation at 37ºC. Sample was collected at set time point of 0.5, 1.0, 2.0 hours, 4.0 hours, and 8.0 hours, respectively. The cells in the sample were decomposed by protein lysate on ice for 45 min and were centrifuged at 12000 rpm under 4? for 10 min. Acetonitrile/methanol (1:2 in V:V) for protein sedimentation was added at 1:1 volume in the supernatant and the mixture was centrifuged at 12000 rpm for 10 minutes. Then, the supernatant was submitted to detect the content of Pcb, which was defined as Pcb content inside cells (Cinside). Furthermore, the supernatant of the cell culture medium was also treated with acetonitrile/methanol (1:2) for protein sedimentation at 1:1 volume and then the mixture was centrifuged at 12000 rpm under 4 ? for 10 minutes. Afterwards, the supernatant was also submitted to detect the Pcb content, which was defined as Pcb content outside cells (Coutside). Here, High Performance Liquid Chromatography (HPLC) method was employed to detect the Pcb content; where the peak area of Pcb at the beginning was set as 100%, which was represented as C0. The transformation percentage of Pcb was calculated as following:
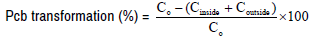
Determination of tyrosinase activity
In a 6-well plate, B16/F10 cells (2×105 cells/ml) were seeded. Collection of the cells were carried on and then washed with PBS after the treatment of Pcb, NAT, and Pcb/NAT (1:1 mole ratio) according to the set concentrations (25 μM). Then, tyrosinase extract buffer was applied to lyse the cells by sonicator for 30 s, and the mixture was placed on ice for 45 min. Then, it was centrifuged at 12000 g under 4? for 15 min. Using a BCA protein assay kit, the protein concentration was determined. By using tyrosinase activity kit (Solarbio, China) detected at an excitation wavelength of 475 nm, the enzyme activity of the extraction was obtained.
Detection of the in vivo antitumor activity
Male C57BL/6 mice (6-8 weeks, body weight: 18 g-22 g) were randomized into 3 groups, which were labeled as A to C groups (n=8). B16/ F10 cells (1 × 107 cells/mouse) were subcutaneously injected into the armpit of the right hind limb of the animals. Group A was set as the tumor control (vehicle group), which received only sodium suspension (0.9%); Group B was set as Pcb group (20 mg/kg), and group C as Pcb/NAT combination group, where the combination was Pcb (20 mg/kg)+NAT group (20 mg/kg). Once daily for 14 days, the animals were injected with either Pcb or Pcb/ NAT into their abdominal cavity. Twenty-four hours after the last dose, blood was collected from the animals for the detection of White Blood Cell (WBC), where retro-orbital puncture under slight anesthesia (diethyl ether) assay was used. At the end of the experiments, the mice were anesthetized by i.p. injection of 5 ml/kg of 1% pentobarbital sodium salt. Under anesthesia, all mice were sacrificed via euthanasia. Weights of each mouse and the separated tumor were determined and recorded. By tumor weighing and tumor-volume measuring, antitumor activity was calculated. Calculation of the tumor inhibition rate was shown below: Tumor inhibitory rate (%) = (1-average tumor weight of administration group/average tumor weight of the control group) × 100%. The thymus and spleen were simultaneously weighed and excised. Calculation of indices were shown below: Index of thymus=[weight of the thymus (mg) × 10]/weight of body (g); Index of spleen=[weight of the spleen (mg) × 10]/weight of body (g).
Analysis of western blotting
Collection of B16/F10 cells was pursued on following PBS wash after the treatment of Pcb, NAT, and Pcb/NAT (1:1 mol ratio), respectively, in accordance with the set concentration (25 μm). Then, RIPA buffer was added to treat the cells by sonication for 30 s and the mixture was placed on ice for 45 min; In the case of tumor, firstly tumor was taken out from the mice, then it was extracted with RIPA buffer 1:10 according to the tumor weight by tissue homogenizer (IKA, Germany). Then centrifugation of the mixture was carried on at 12000 g under 4? for 15 min. Collection of the supernatants was undergone and it was submitted for determination of BCA protein and protein analysis by SDS-PAGE method. Using the nucleoprotein extraction kit (Beyotime, Shanghai, China), nuclear protein of the cells was obtained. In order to denature, loading buffer was added to the nucleoprotein extraction and the mixture was maintained at 100°C for 5 min. By using SDS-PAGE method, proteins were separated and submitted to 0.45 μm PVDF (polyvinylidene fluoride) membranes. Blockade of the membranes was carried on in 5% skim milk for 2 h at room temperature following incubation with primary and secondary antibodies. The primary antibodies were rabbit antibodies for β-Actin (1:3000), Tyrosinase (1:2000), GAPDH (1:3000), p53 (1:2000), Bcl-2 (1:1000), Bax (1:1000) in TBST buffer, respectively. They were incubated with membranes at 4°C overnight, respectively. The secondary antibody was the HRP-conjugated goat anti-rabbit IgG diluted at 1:3000 in TBST buffer. After the membranes were incubated with super ECL plus detection reagent (Applygen, Beijing, China), protein bands were identified by chemiluminescence. Quantification of protein bands were done by the Image J software, reference protein was β-actin. Through normalization to β-actin, the densities of each band were obtained. Based on these normalized densities, the ratio of different proteins was calculated.
Analysis of statistics
Mean±standard deviation (SD) of three independent experiments is expressed as the forms of all data. One-way ANOVA (for comparisons of three or more groups) was applied to assess statistical significance. Statistically significance was defined when P-value <0.05.
Pcb/NAT treatment enhanced the inhibitory sensitivity against B16/F10 and K1735 cells
Since tyrosinase is highly expressed in the murine melanoma B16/F10 cell, it was employed as model cell herein. In order to determine the effects of Pcb and different Pcb/NAT combination on the cellular growth inhibition, MTT assay was used. As displayed in Figure 1A, combination of Pcb with NAT (Pcb/NAT) in 1:1 mole ratio made the inhibition activity of Pcb against B16/F10 cells increase from IC50 value of 31.9±1.1 to 14.2 ± 1.1 μM, which was 2.24 times better than Pcb solely. The combination of Pcb/NAT in 1:1 mole ratio showed the best activity after the comparison with other ratio of 3:1, 2:1, 1:2, 1:3, and 1:4 (data not shown here).

Figure 1: Cytotocicity comparison of Pcb, NAT, and Pcb/NAT combination against B16/F10 and K1735 cancer cell lines and morphologic changes. (A) B16/F10, and K1735 (B) cells were treated with Pcb, NAT, and Pcb/NAT combination for at 37°C for 48 h at different concentration, respectively. Cell viability was determined by MTT assay. Each experiment was repeated 3 times. The results were expressed as mean±standard error of the mean (SD); **P<0.01 vs Control group, ##P<0.01 vs Pcb-treated group. (C-E) B16/F10 cells were treated without (C) or with 25 μM Pcb (D), and Pcb/NAT (E), respectively at 37°C for 48 h. Representative cell morphology was visualized by a fluorescence microscope with (40×), and (100×), respectively. The images shown are representative of three experiments; (F) The growth curve of B16/F10 cancer cell line. B16/F10 cells were treated without or with Pcb or Pcb/NAT at a time duration of 60 h. The cell viability of B16 cells were measure using MTT assay at setting time point. Results are shown as the mean±SD. *p<0.05, **p<0.01 vs Control group; #p<0.05, ##p<0.01 vs. Pcb group.
To get rid of the possibility that this difference is because of the B16/F10 cell line-specific, another mouse melanoma cell line K1735 was applied. It was found that group Pcb/NAT raised the IC50 value from 43.8 ± 1.3 μM to 16.3 ± 0.9 μm (Figure S5).
In order to clarify the role of NAT, we investigated the cytotoxicity of NAT at different concentration. It was found that under 20 μm, treatment of NAT slightly promoted the proliferation of both cells (B16/F10 and K1735). Within 50 μm, NAT showed nontoxic to both cells (Figures 1A-1B).
All these evidences implied that combination of NAT enhanced the inhibition sensitivity of Pcb against B16/F10 and K1735 melanoma cells.
Pcb/NAT treatment made B16/F10 cells differentiate into normal cells better than Pcb
As we know, tumor formation is related to abnormal cell differentiation. Usually, tumor cells can not differentiate normally or have very low differentiation ability [15]. After the tumor cells are damaged, there is a tendency to induce the differentiation of the tumor cells into normal cells. It has been reported that B16/F10 cells has typical dendritic processes in morphology when differentiating into normal cells [16, 17].
The results of cell morphology shown in Figure 1C indicated that the normal B16/F10 cells were long spindle with few edges, small cell gap and high cell density. Interestingly, after Pcb-treatment, the volume of cells increased in a shape of star polygon with dendritic structure (Figure 1D). Compared to Pcb group, treatment of Pcb/NAT combination resulted in lower cell density and more obvious differentiation (Figure 1E), which implied that Pcb/NAT combination had better inhibitory effect against B16/ F10 cell growth.
To further investigate the inhibitory effect, a growth cure analysis was carried on (Figure 1F). It was shown that Pcb itself inhibited cell proliferation in a time-dependent manner. Interestingly, without no doubt, the inhibitory effect of Pcb/NAT was more obvious than that of Pcb solely.
Pcb/NAT treatment caused more cell apoptosis
Concluding above, Pcb/NAT combination displayed more sensitive inhibition against B16/F10 cells than Pcb. Therefore, we would like to make a profound study to disclose the anticancer mechanism involved in the combination. Firstly, cell apoptosis was investigated. In this experiment, propidium iodide (PI) and Annexin-V were used as dyes to label viable and dead cells, flow cytometry was applied for the detection.
As displayed in Figure 2, compared to control group (Figure 2A), Pcb significantly promoted cell apoptosis. Pcb at 25 μm induced about 45% total apoptosis rate, in which 41% was for late apoptosis and 4% for early (Figure 2B). Interestingly, the combination at the same dose induced higher apoptosis rate with about 65% for total, where 60% for late and 5% for early (Figure 2C). Figure 2D demonstrates the comparison of apoptosis rate in B16/F10 cells between Pcb and Pcb/NAT treatment. Clearly, addition of NAT resulted in more cell death in late apoptosis stage.

Figure 2: Representative scatter diagrams of Pcb and Pcb/NAT combination induced apoptosis in B16/F10 cells. B16 cells were pre-treated without (A) or with 25 μM Pcb (B), and Pcb/NAT (C), respectively at 37°C for 48 h. Cells were stained with Annexin-V and PI. The apoptosis of B16 cells was detected by flow cytometry. The evaluation of apoptosis is via Annexin V: FITC Apoptosis Detection Kit per manufacture’s protocol. In each scatter diagrams, the abscissa represents the fluorescence intensity of the cells dyed by Annexin V; and the ordinate represents the fluorescence intensity of the cells dyed by PI. The lower left quadrant shows the viable cells, the upper left shows necrotic cells, the lower right shows the early apoptotic cells; while the upper right shows late apoptotic cells. (D) The histogram of cell apoptosis rate in each group. Cell apoptosis rate is defined as (NR2+NR3+NR5)/(NR2+NR3+ NR4+NR5)×100%, N means cell population in the quadrant; **P<0.01, ***P<0.001 vs Control group.
Pcb/NAT treatment decreased more mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential (ΔΨm) decline is an early marker of apoptosis. Here, we used JC-1 as a fluorescent probe to detect ΔΨm. It was clearly disclosed in Figures 3A-3D that the ΔΨm of B16/F10 cells decreased by Pcb from 0.23 in control group to 0.19. Quite interestingly, involvement of NAT further reduced ΔΨm by 21%, which was 0.15 (Figure 3D). This was consistent with the fact that the combination of NAT increased late cell apoptosis induced by Pcb.

Figure 3: Flow cytometry analysis of cell mitochondrial membrane potential (ΔΨm) based on JC-1 staining in B16/F10 cells treated by Pcb and Pcb/NAT. B16 cells were pre-treated without (A) or with 25 μm Pcb (B), and Pcb/NAT (C), respectively at 37°C for 48 h. Then, the cells were collected and washed twice with PBS. After adding 0.5 mL JC-1 solution and incubating at 37ºC for 30 minutes, the mixture was centrifuged at 800 rpm, washed three times with JC-1 staining buffer. The membrane potency of B16/F10 cells was detected by flow cytometry and the data were evaluated using the Flowjo 7.6.1 software. (D) The histogram of cell mitochondrial membrane potency in each group. The results represent mean ± SD of three separate experiments,*P<0.05, **P<0.01 vs. Control group.
Pcb/NAT treatment arrested more cells at G0/G1 phase
To establish whether the inhibition of Pcb and Pcb/NAT against cell growth is related with the cell cycle progress, analysis of cellular DNA was carried on after propidium iodide (PI) staining. Flow cytometry was used for this analysis. The profiles were displayed in Figures 4A-4D. Apparently, compared to the control group, an increase in the G0/G1 population was found in B16/F10 cells while the S and G2/M population were down after the treatment of Pcb, and Pcb/NAT at 25 μm, respectively. Pcb/NAT combination arrested more cells at G0/G1 state than Pcb solely. It up-regulated G0/G1 population by 7.8% but decreased by 25.7% in G2/M state compared to the Pcb group.

Figure 4: Cell cycle analysis of B16/F10 cells exposed to Pcb and Pcb/NAT combination, respectively. B16 cells were pre-treated without (A) or with 25 μm Pcb (B), and Pcb/NAT (C), respectively at 37°C for 48 h. Cells were collected, fixed in 70% ethanol, and stained with propidium iodide solution. G0/G1: quiescent state/growth phase; S: initiation of DNA replication; G2/M: biosynthesis/mitosis phases. (D) The percentage histogram of cell arrested in each group. *p<0.05, **p<0.01 vs Control group.
Combination of NAT promoted the transformation of Pcb in B16/F10 cells
It had already been confirmed that in vitro, tyrosinase promoted the transformation of Pcb at the presence of NAT through the indirect oxidation. In the current investigation, we would like to verify that combination of NAT might promote the transformation of Pcb in B16/F10 cells. As indicated in Figure 5A, Addition of NAT sped up the transformation of Pcb in B16/F10 cells. After 8 hours incubation, the transformation rate in Pcb group was 30%; while in Pcb/NAT group, it was 45%. This indicated that the existence of NAT accelerated Pcb transformation.

Figure 5: Transformation percentage of Pcb, tyrosinase activity, and Western blot analysis of tyrosinase and GAPDH in B16/F10 cells. (A) Transformation percentage of of Pcb. B16 cells were treated with 25 μm of Pcb, and Pcb/NAT (1:1 mol ratio), respectively in a procedure of 24-hours incubation at 37ºC. Sample was collected at set time point of 0.5, 1.0, 2.0, 4.0, and 8.0 h, respectively. The content of Pcb was detected by HPLC method; (B) Activity of tyrosinase. B16/F10 cells were collected and washed with PBS after the treatment with 25 μM of Pcb, NAT, and Pcb/NAT (1:1 mol ratio), respectively. The protein concentration was measured using a BCA protein assay kit. The extraction was tested by tyrosinase activity kit (Solarbio, China) with an excitation wavelength at 475 nm; Protein expressions of tyrosinase (C) and GAPDH (D) in B16/F10 cells after the treatment of 25 μm of Pcb, NAT, and Pcb/NAT (1:1 mol ratio), respectively using Western blot. The density of each lane was presented as mean ± standard deviation (SD) for at least three individual experiments. Blots were quantified using Image J software. *P<0.05, **P<0.01 vs. Control group; (E) Possible mechanism for the transformation of Pcb at the presence of NAT.
Pcb/NAT treatment increased more the expression and activity levels of tyrosinase and GAPDH in B16/F10 cells
According to the previous studies, the bio-activation of Pcb can be postulated to proceed through azoprocarbazine and the azoxyprocarbazine isomers. These intermediates were subsequently transformed into methyl radical or other radicals, which were charged for anticancer activity of Pcb. In order to explore the transformation mechanism, we detected the activity and expression of related proteins.
Concluding from Figure 5B, tyrosinase activity was significantly increased after treated by Pcb, and NAT, respectively (P<0.05); NAT activated tyrosinase a little bit more than Pcb. However, Pcb/NAT combination synergistically enhanced the enzyme activity, which was 1.21 times fold more than Pcb solely (P<0.01). More interestingly, protein expression of tyrosinase in B16/F10 cells was also up-regulated by Pcb, NAT, and Pcb/NAT treatment, respectively. The involvement of NAT with Pcb also synergistically increased the protein expression to a greater degree, which was 1.2 times fold than Pcb itself (Figure 5C).
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) may be related to the hydrazines and directly produce azo compounds and free radicals [18]. To investigated the effect of Pcb and Pcb/NAT on GAPDH protein level, B16/F10 cells were treated with Pcb or Pcb/NAT, as shown in Figure 5D, the protein expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in B16/F10 cells was influenced by the treatment of Pcb, and Pcb/NAT, respectively. The combination up-regulated the expression level of GAPDH by 1.48 times fold than Pcb solely. Figure 5E displayed a putative mechanism of Pcb transformation.
Pcb/NAT treatment impacted on the apoptosis proteins expression more
Several factors, including the expression of B-cell lymphoma 2 (Bcl- 2) family members such as Bcl-2, and Bcl-2 associated protein X (Bax), can trigger mitochondrial-mediated apoptotic pathway. The permeability of the mitochondrial membrane can be affected by this protein family leading to the release of cytochrome c and mitochondrial dysfunction, finally cell apoptosis [19]. Herein, anti-apoptotic protein Bcl-2 was found down-regulated after the treatment of Pcb, and Pcb/NAT combination at 25 μM, respectively (Figure 6A). Usually, when Bcl-2/Bax ratio becomes less, percentage of cell apoptosis increases. It was found that Pcb/NAT decreased the Bcl-2/Bax ratio to 0.7, which was far less than Pcb group (1.1) and the control group (1.2) (Figure S6).

Figure 6: Western blot analysis of Bcl-2/Bax and p53 in B16/F10 cells, antitumor activity and Western blot analysis of tyrosinase and GAPDH in B16/F10-bearing mice. Protein expressions of Bcl-2/Bax (A) and p53 (B) in B16/F10 cells. After the treatment of 25 μm of Pcb, and Pcb/NAT (1:1 mol ratio), respectively, using western blot. The density of each lane was presented as mean±standard deviation (SD) for at least three individual experiments. Blots were quantified using Image J software. *P<0.05 vs. Control group; Antitumor activity (C), the growth inhibition rate of tumor volume (D) and protein expressions of tyrosinase (E) and GAPDH (F) in B16/F10-bearing mice. Control: Vehicle; Pcb: 20 mg/kg Pcb; Pcb/NAT: 20 mg/kg Pcb/NAT (1:1 mol ratio). *p<0.05, **p<0.01 vs. control group.
p53 is known as a tumour suppressor which takes part in many roles such as induction of cell cycle arrest, DNA repair, senescence, and apoptosis [20,21]. Excitingly, Pcb and Pcb/NAT combination were shown to up-regulate p53 expression, respectively (Figure 6B). It was obvious that Pcb/NAT has more influence on the regulation of p53 than Pcb solely.
Pcb/NAT treatment enhanced tumor inhibition in vivo
In order to evaluate the antitumor activity of Pcb and Pcb/NAT in vivo, B16/F10-bearing mice model was set up. As shown in Figures 6C-6D and Table 1, Pcb /NAT combination showed better tumor inhibition than Pcb itself, where the tumor inhibition rate was 89.4% for the combination group, and 84.5% for Pcb (Table 1). Moreover, a tumor inhibition analysis based on tumor volume found that Pcb/NAT combination showed better tumor inhibition than Pcb itself, where the tumor growth inhibition raised from 86.8% to 95.6% (Figure 6D).
| Group | dosage (mg/kg) | Body weight (g) | Tumor weight (g) | Inhibitory rate (%) | Thymus index | Spleen index |
|---|---|---|---|---|---|---|
| Vehicle | — | 32.2 ± 1.6 | 0.66 ± 0.52 | — | 1.09 ± 0.52 | 3.83 ± 1.04 |
| Pcb | 20 | 31.1 ± 2.0 | 0.10 ± 0.12* | 84. 5 | 1.32 ± 0.59 | 4.13 ± 0.67 |
| Pcb/NAT | 20 | 31.3 ± 2.3 | 0.07 ± 0.05* | 89.4 | 1.25 ± 0.35 | 4.64 ± 1.15 |
“—” means no data; Values were expressed as mean ± SD (n=6); Significance was determined using one-way ANOVA, *p<0.05 vs. vehicle group.
Table 1: Effects of Pcb and Pcb/NAT on body weight, tumor weight, tumor inhibitory rate and immune organ index of B16/F10-bearing mice.
Of notice, i.p. administration of either Pcb or Pcb/NAT combination caused a little bit loss of body weight, and an increase in both indexes of spleen and thymus in tumor-bearing mice (Table 1). We found that Pcb/ NAT showed a little less in thymus index, while a little more in spleen index than Pcb. As we know, changes of either thymus index or spleen index are a close indication of the immune status in life. Upon this view point, either Pcb or Pcb/NAT might have an impact on immune system.
Furthermore, in Table 2, it was found that Pcb-treated groups significantly cut down the level of platelet (PLT) (P< 0.05 vs vehicle group). Interestingly, with the involvement of NAT, either WBC level or PLT was raised a little bit.
| Group | dosage (mg/kg) | WBC (103/μl) | PLT (103/μl) | RBC (106/μl) | HGB (g/dL) |
|---|---|---|---|---|---|
| Vehicle | — | 16.4 ± 14.4 | 174.2 ± 88.4 | 9.24 ± 8.96 | 15.35 ± 0.67 |
| Pcb | 20 | 9.4 ± 4.2 | 77.2 ± 26.1* | 9.60 ± 9.81 | 14.24 ± 1.64 |
| Pcb/NAT | 20 | 9.9 ± 6.5 | 82.7 ± 39.6* | 9.80 ± 9.23 | 14.66 ± 0.94 |
“—” means no data; Values were expressed as mean ± SD (n=6); Significance was determined using one-way ANOVA; *p<0.05 vs. vehicle group.
Table 2: Effects of Pcb and Pcb/NAT on WBC, RBC, HGB, and PLT of B16//F10-bearing mice.
Pcb/NAT treatment up-regulated in vivo expression of ty - rosinase and GAPDH
To confirm whether tyrosinase and GAPDH were influenced in solid tumor, we tested their protein expression by Western blot. The result in Figure 6E showed that both Pcb (p<0.05) and Pcb/NAT (p<0.01) groups upregulated tyrosinase expression. Of notice, the latter group had a greater influence than Pcb group by 1.37 folds. Interestingly, treatment of Pcb alone did not increased GAPDH expression (Figure 6F). However, Pcb/NAT did upregulate GAPDH expression in B16/F10-bearing tumor (p<0.05) (Figure S7).
Generally, melanoma is considered as a complex genetic disease. The loss of adhesion receptors, mutations in critical growth regulatory genes, and the production of autocrine growth factors, all these gene dysfunctions may probably disrupt intracellular signaling in melanocytes, resulted in the loss of tight regulation by keratinocytes [22]. Melanocytes originate from highly motile cells; this probably enhances the survival properties of the melanoma cells. This is the reason why melanoma is an extremely aggressive disease. It shows notoriously high resistance to cytotoxic agents and high metastatic potential. Compared with other tumor cell types, melanoma cells display low levels of in vivo spontaneous apoptosis and relatively in vitro resistant to drug-induced apoptosis [23].
As a part of a quadruple drug regimen in the treatment of Hodgkin’s disease, Pcb had ever been used as a single agent to treat melanoma with 28% response rate and prolonged duration of response in a collected small series [24]. This drug, with hydrazine structural moiety, is normally converted to its active forms by oxidative enzymes inside the organism. Excitingly, we happened to learn that tyrosinase may indirectly transform Pcb to its bioactive entities at the present of phenolic compounds; Therefore, we realized at once that this must be a way to enhance the anticancer sensitivity of Pcb against melanoma.
As we know, in melanogenesis, tyrosinase catalyzes the initial hydroxylation of L-tyrosine to L-Dopa and the rate limiting step, which is the subsequent oxidation to dopaquinone leading to melanin [25]. This was the reason why tyrosinase was considered long time ago as a target for chemotherapy of melanoma [26-28]. Unfortunately, most of them including the first candidate 4-hydroxyanisole, which reached clinical trials, were confirmed with significant renal and hepatotoxicity [29]. Later, another strategy, which was named as Melanocyte-directed Enzyme Prodrug Therapy (MDEPT), has drawn substantial attention [30,31]. However, irrationality in drug structure results in unlikely release of active anti-tumor entity [29]. No doubt, this MDEPT strategy needs to be further improved.
We suggested that combination of an anti-melanoma hydrazine agent with a safe quinone-directed tyrosinase substrate may be an effective way to enhance the anti-cancer activity. This was supported by our investigation. In the current study, the Pcb/NAT did increase the inhibition sensitivity of Pcb against B16/F10 cells (Figure 1A). Combination of NAT sped up the transformation of Pcb to azo-procarbazine (Figure 5A) based on the mechanism indicated in Figure 5E. With the present of NAT, there are two ways to realize the transformation. One is the original oxidation of Pcb primarily by cytochrome P450 monooxygenases and peroxidases [11]; another is the participation of tyrosinase. This enzyme changes NAT into N-acetyl-L-dopaquinone, which may dehydrogenize Pcb into azoprocarbazine (Figure 5E) [12].
Pcb alone was shown the in vitro and in vivo ability to increase the tyrosinase amount and activity (Figures 5B and 5C, Figure 6E), and induce B16/F10 melanoma cell differentiation (Figure 1D). These functions are similar to that of PD 98059, which is a specific inhibitor against mitogenactivated protein (MAP) kinase kinase (MEK) [30]. As we know, MEK is involved in Ras/Raf/MEK/ERK pathway, which is a key regulator of melanoma cell proliferation, with ERK being hyperactivated in up to 90% of human melanomas [31]. However, whether Pcb inhibits or blocks the MAP kinase pathway needs to be further investigated. Interestingly, involvement of NAT with Pcb strengthened all the tendencies induced by Pcb solely. We confirmed that NAT alone increased tyrosinase activity and protein level in vitro. However, Pcb/NAT had a synergistic effect.
It was shown clearly that glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was involved in the anti-melanoma process of Pcb. Treatment of Pcb increased the protein expression in B16/F10 cells (Figure 5D), but this is not significant in B16/F10-bearing tumor (Figure 6F); However, the combination of NAT with Pcb enhanced GAPDH protein level in vitro and in vivo.
Traditionally, GAPDH was considered a classical glycolytic protein for its pivotal role in energy production. Quite interestingly, afterwards, there are many evidences demonstrating that mammalian GAPDH displays a number of diverse activities such as microtubule bundling, phosphotransferase activity, nuclear RNA export, DNA replication, DNA repair, and its role in membrane fusion. GAPDH might play a role in prostate cancer concluded from its high mRNA levels in both highly metastatic rat adenocarcinoma cell lines and neoplastic tissues [32,33]. The compensate biosynthesis of GAPDH will be initiated during programmed cell death [23,34]. Therefore, we suggested that the amount increase of GAPDH in the current investigation was closely related to the compensate biosynthesis caused by cell apoptosis induced by either Pcb or Pcb/NAT (Table S1).
Acetaldehyde was found to be oxidized to acetyl phosphate by GAPDH through dehydrogenation [35]; however, whether GAPDH takes part in the dehydrogenation of Pcb to azoprocarbazine remains unknown. As we know, GAPDH, together with α-glycerolphosphate dehydrogenase (αGPDH) are primarily associated with pre-meiotic germ cells. After a single i.p. injection in (C57BL/6 × DBA/2) F1 male mice, Pcb was found to cause dosedependent decreases in sperm count [2]; In the meantime, it also induced in situ activity increase of αGPDH and GAPDH [29,36].
Basically, the generation of alkyl radicals and alkylation under some enzymatic systems were recognized as the ways for Pcb to function in both antineoplastic action and carcinogenesis [2]. However, Pcb was also found to cause DNA damage non-enzymatically [37]. This is in consistent with the evidences that both Pcb and Pcb/NAT arrested more cells at G0/G1 phase (Figure 4) and up-regulated p53 expression (Figure 6B). We postulated that in response to DNA damage, p53 is most likely activated and the transcription of p21 (WAF1, Cip-1) is then turned on [38], this downstream gene acts on cyclin–Cdk complexes and PCNA to stop DNA replication, thus enforces G1 arrest [39] (Table S2).
The decision to commit to an apoptotic Bax gene may be influenced by p53 [40]. Binding of Bax to Bcl-2 antagonizes its ability to block apoptosis. Over-expression of Bcl-2 will block p53-mediated apoptosis. In our study, we confirmed that both Pcb and the combination activated p53, resulted in the decrease of Bax/Bcl-2 ratio (Figure 6A), therefore increased B16/ tendencies induced by Pcb without bringing about toxicity increase in vivo (Tables 1 and 2).
In a word, this study showed that the Pcb/NAT combination enhanced the anti-melanoma sensitivity of Pcb. In all cases, combination of NAT strengthened all tendencies induced by Pcb but no increase in toxicity. All evidences suggest that Pcb/NAT is a promising anti-melanoma agent to replace Pcb.
Medicinal Chemistry received 6627 citations as per Google Scholar report



