Research Article - (2024) Volume 20, Issue 1
Received: 01-Jan-2024, Manuscript No. JOS-23-124774;
Editor assigned: 02-Jan-2024, Pre QC No. P-124774;
Reviewed: 17-Jan-2024, QC No. Q-124774;
Revised: 23-Jan-2024, Manuscript No. R-124774;
Published:
31-Jan-2024
, DOI: 10.37421/1584-9341.2024.20.126
Citation: Sonwani, Hari Prasad. “NVP-231, a Ceramide Kinase Inhibitor, Causes M Phase Arrest and Consequent Cell Death to Stop the Growth of Breast and Lung Cancer Cells.” J Surg 20 (2024): 126.
Copyright: © 2024 Sonwani HP. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Context and objective: The production of ceramide-1-phosphate, which is catalysed by ceramide kinase (CerK), may influence a number of cellular processes, including inflammation and cell division. Here, we looked at how the newly created CerK inhibitor NVP-231 affected the viability and proliferation of cancer cells, as well as the function of cell cycle regulators linked to these reactions.
Experimental methodology: Gradually increasing concentrations of NVP-231 were applied to the lung cancer cell line NCI-H358 and the breast cancer cell line MCF-7 in order to measure DNA synthesis, colony formation, and cell death. The cell cycle distribution of the cells was analyzed using flow cytometry, and alterations in the expression and activation of cell cycle regulators were found using Western blot analysis.
Important findings: NVP-231 decreased DNA synthesis, colony formation, and cell survival in both cell lines in a concentration-dependent manner. Additionally, it caused apoptosis, as seen by elevated caspase-3 and caspase-9 cleavage and DNA fragmentation. NVP-231 reduced the proportion of cells in the S phase and caused an M phase arrest with an elevated mitotic index, as shown by increased histone H3 phosphorylation, according to cell cycle analysis. When staurosporine was added to NVP-231 therapy, the impact on the cell cycle became even more noticeable. Lastly, siRNA-induced down-regulation of CerK sensitized cells to staurosporine-induced apoptosis, although overexpression of CerK protected against this process.
Conclusions and significance: our findings reveal for the first time the critical function that CerK plays in the regulation of the M phase in cancer cells. We propose that CerK be specifically inhibited, with the use of medications like NVP-231, in conjunction with standard pro-apoptotic treatment.
Fragmentation • Staurosporine • Caspase-3 and caspase-9 • Lung cancer cell line NCI-H358
BrdU: 5-Bromo-2-deoxyuridine; C1P: Ceramide-1-Phosphate; CerK: Ceramide Kinase; CDK: Cyclin-Dependent Kinase; NVP- 231: N-[2-benzoylamino-1, 3-benzothiazol-6-yl] Adamantane-1-Carboxamide; NVP-995: N-[2-[(3, 4-Dimethoxybenzoyl) Amino]-1, 3-Benzothiazol- 6-yl] Adamantane-1-Carboxamide; S1P: Sphingosine-1-Phosphate
While sphingolipids are crucial for the structural integrity of cellular membranes, it has also been demonstrated that some of its subspecies function as signaling molecules. Numerous investigations have demonstrated their crucial function in controlling cell viability and division. Over the past few decades, a great deal of research has been done on the functions of ceramide and Sphingosine 1-Phosphate (S1P) in relation to cell growth and death. S1P appears to be a countermolecule to ceramide, as it appears to have opposite effects in many cell types, such as promoting cell survival and proliferation [1-4]. Ceramide's effects are ant proliferative and pro-apoptotic. Ceramide 1-Phosphate (C1P), a different type of phosphorylated sphingolipid, has garnered attention recently because it may regulate a number of cellular processes [5]. Functions include the release of synaptic vesicles, phagocytosis, mast cell degranulation, inflammatory reactions, proliferation, and angiogenesis. These are just a few examples of the functions that are described in the literature. While Gomez-Muñoz A, et al. [6], suggested that C1P could induce cell migration and proliferation, this effect was primarily observed when exogenous C1P was added to cell cultures. It was suggested that the migratory impact of exogenous C1P operates via a putative C1P receptor that exhibited sensitivity to the pertussis toxin. Nevertheless, extremely high C1P concentrations between 30 and 50 μM were required for this, indicating that if a receptor is engaged, there is little affinity for interaction. There isn't yet a known high-affinity C1P receptor. Ceramide kinase (CerK), which co-purified with synaptic vesicle markers in brain synaptic vesicles, is responsible for the generation of C1P. In the meantime, clones of CerK have been found in mice and humans. Sugiura M, et al. [7] reported that CerK is a 537 amino acid protein that exhibits similarities to both DAG kinase and sphingosine kinase 1. However, it demonstrates significant substrate specificity for ceramides, particularly the short-chain analogues C6-, C8-, and C16-ceramide. CerK has been demonstrated to be distributed throughout the cytoplasm, nucleus, and Golgi. The order of the N-terminus of According to Carré A, et al. [8], CerK is believed to be myristoylated and to have a pleckstrin homology domain, which is crucial for Golgi localization. Additionally, it was demonstrated that a nuclear export signal is present in the C-terminal region and a nuclear import signal is present in the N-terminal area. Based on these data, a novel regulatory mechanism involving the nucleocytoplasmic shuttling of CerK was postulated. Considering that no C1P transporter has been found to date, it makes sense that the primary location of C1P synthesis is inside the cell and, by extension, the principal site of action may likewise be in the intracellular region, given these reports. In this study, we looked into the impact of a freshly created catalytic inhibitor of CerK NVP-231 on the proliferation and survival of the lung and breast cancer cell lines NCI-H358 and MCF-7. We showed that this inhibitor decreases DNA synthesis, colony formation, and cell viability in a concentration-dependent manner. Additionally, we showed that cells arrest in the M phase of the cell cycle and then go through apoptosis. The overexpression of CerK in the cells decreased the death-promoting effect of NVP-231. Moreover, staurosporine and NVP-231 together had a synergistic effect on the activation of apoptosis. These results highlight the critical function that CerK plays in the regulation of mitosis and indicate that pro-apoptotic anticancer drugs should be used in combination with targeted CerK inhibition as a therapeutic approach.
Culture and stimulation of cells
The American Type Culture Collection provided the human lung cancer cell line NCI-H358 and the human breast cancer cell line MCF-7. NCI-H358 cells were grown in accordance with earlier instructions. In addition to 10% (v·V-1) FBS, 10 mM HEPES pH 7.4, 6 μg·mL−1 insulin, 100 units·mL−1 penicillin, 100 μg·mL−1 streptomycin, and non-essential amino acids, MCF-7 cells were grown in DMEM. Cells were cultivated at 37 ℃ in a humidified environment with 5% (v·V-1) CO2 throughout all experiments. After homogenizing stimulated cells in lysis buffer, they were centrifuged at 13,000 × g for 10 minutes [9]. 30 μg of protein were isolated from the supernatant after it was used for protein determination. By SDS-PAGE, transferred to nitrocellulose membrane, and then, using the antibodies listed in the figure legends, submitted to Western blot analysis as previously described.
Transfection of cells
After a day, 4 × 105 MCF-7 cells per well were implanted in 6-well plates until they were 50–70% confluent, ensuring persistent CerK overexpression. The cells were subsequently cultured for a full day in serum-free DMEM supplemented with a combination of 4 μL·mL−1 of TurboFectR transfection reagent (Fermentas GmbH, St. Leon-Rot, Germany) and 2 μg·mL−1 plasmid DNA (pcDNA3.1 plus human CerK cDNA). TurboFectR and the empty pcDNA3.1 vector were used to transfect control cells. Before conducting tests, transfected cells were kept for a minimum of three weeks in full media containing 1 mg mL−1 G418 for selection. According to the manufacturer's instructions, cells were transfected with oligofectamine and 100 nM of a Smart pool siRNA of hCerK (Dharmacon RNAi, L-004061; Fisher Scientific, Wohlen, Switzerland) in order to downregulate CerK. Fourteen hours later For the DNA fragmentation test, cells were subcultured into 96-well plates following transfection. Cells were treated in accordance with the figure legends after a 24-hour period.
Assay for cellular CerK activation
The procedure for performing the cellular CerK activity assay was followed by Boath A. Using the Prism 5.03 program (GraphPad Software Inc., La Jolla, CA, USA) and a non-linear curve fit, the cellular IC50 value was determined.
Assay for DNA synthesis and proliferation
After plating 1 × 104 cells per well in a 96-well plate, the cells were kept in growth media for the whole night. Following the recommended course of treatment, 5-Bromo-2-deoxyuridine (BrdU) was added to the culture media for the final 24 hours. Following the manufacturer's recommendations, incorporated BrdU was found using the Cell Proliferation BrdU elisa kit (Roche Diagnostics GmbH, Mannheim, Germany).
Alamar blue assay for cell viability
The Alamar Blue R reagent (reassuring) from Life Technologies, Thermos Fisher Scientific, Waltham, MA, USA, was used to evaluate the vitality of the cells. After plating cells in a black 96-well plate with 7.5 × 103 cells per well, the cells were kept in full media for the whole night. After that, the cells were treated as directed, and the alamar Blue reagent was added for the final four hours. Using a SpectraMax microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA), the intensity of the fluorescence representing cell viability was evaluated at 544/590 nm excitation/emission wavelengths. Alternatively, flow cytometry was used to find the vitality of the cells. In order to do this, treated cells were trypsin zed, once again cleaned in PBS, and then suspended in PBS with 10 μg·mL−1 propodeum iodide and 1% FCS. Following a minute of incubation at 25 ℃, cells were examined right away. using flow cytometry. Using a FACSCalibur (Becton & Dickinson, San Jose, CA, USA), propodeum iodide (PI)-positive staining was found in the FL3 channel. Each sample had at least 20,000 cells counted.
Assay for colony development
In dishes with a diameter of 60 mm, 1000 cells were cultivated per dish. To allow for colony formation, cells were cultured for an additional 10 days (NCI-H358) or 14 days (MCF-7) after being treated as directed in the growth medium for 24 hours. After that, the cells were air dried, cleaned with PBS, and stained with 2% (wv−1) crystal violet for half an hour. Using a ColCountTM (Mammalian Cell Colony Counter, Oxford Optronix, Oxford, UK), the number of cell colonies was determined. For counting purposes, only colonies with more than 50 cells were considered.
Flow cytometry analysis of the cell cycle
After being separated by trypsin/EDTA, stimulated cells were preserved in 70% (v·v−1) ethanol for a minimum of one hour at -20 ℃. Following a 30-minute incubation period in PBS containing 10 μg·mL−1 propidium iodide and 100 μg·mL−1 RNase A, the cells were examined using flow cytometry to determine the DNA content using a Becton & Dickinson FACSCalibur. Prior to adding PI to the cell samples, further labelling for the mitotic marker phospho-Ser10 histone H3 was carried out in order to distinguish between cells in the M phase and cells in the G2 phase. Using the FL1 channel and an anti-rabbit Alexa-488 secondary antibody, the phospho-H3 positive cells were identified. For each sample, a minimum of 20,000 cells were counted.
C16-C1P and ceramide quantification using LC-MS/MS C1P and ceramides were extracted and measured in accordance with a previously published protocol [9]. To put it briefly, internal standards C12-C1P and C17- ceramide were used to extract lipids from cells. Using a Q-TOF 6530 mass spectrometer (Agilent Technologies, Waldbronn, Germany) operating in the positive ESI mode for ceramides and the negative ESI mode for C1P, the sample was analyzed using rapid-resolution liquid chromatography-MS/MS. Cleavage of the precursor ions of C12-C1P (m/z 560.408), C16-C1P (m/z 616.471), and ceramides (C16-ceramide (m/z 520.508), C17-ceramide (m/z 534.524), C18-ceramide (m/z 548.540), C18:1-ceramide (m/z 546.524), C20- ceramide (m/z 576.571), C22-ceramide (m/z 604.602), C24-ceramide (m/z 632.634), C24:1-ceramide (m/z 630.618) were split into fragment ions of m/z 78.960, m/z 78.960, and m/z 264.270, respectively. Quantification was carried out utilizing Agilent Technologies' Mass Hunter software.
Analytical statistics
The experimental data was statistically analyzed using unpaired t-tests when only two groups were compared or one-way anova followed by a Bonferroni's post hoc test for multiple comparisons.
Chemicals
Hyperfilm MPR, enhanced chemiluminescence reagents, and secondary HRP-coupled IgGs were obtained from GE Health Care Systems, located in Grautbrugg, Switzerland. Enzo Life Sciences AG (Lausen, Switzerland) was the source of C6-ceramide. Cayman Chemicals Inc. provided the C6-NBDceramide (Ann Arbor, MI, USA) Cell Signaling Technology Europe B.V. (Leiden, the Netherlands) provided the antibodies against GAPDH, total histone H3, cleaved caspase-3, phospho-Ser133-cyclin B1, total cyclin B1, wee1, and cyclin-dependent kinase (CDK)4; phospho-Ser10 histone H3 was from EMD Millipore Inc. (Darmstadt, Germany); cleaved caspase-9 and β-actin were from Santa Cruz Biotechnology, Inc. (Heidelberg, Germany); NVP-231 and N-[2-[(3,4-dimethoxybenzoyl)amino]6-yl-1,3-benzothiazol]Novartis Pharma Inc. (Basel, Switzerland) supplied adamantane-1-carboxamide (NVP-995), whereas Merck Chemicals Ltd. [10] supplied K1. Every additive used in cell culture came from Invitrogen AG, based in Basel, Switzerland.
NVP-231 causes cancer cells to die, which lowers DNA synthesis and cell viability
Graf C., previously reported the new CerK inhibitor NVP-231, which had an IC50 of 12 nM and reduced the catalytic activity of recombinant CerK in vitro. For this reason, this inhibitor is a useful tool in researching CerK's cellular activities. Here, we looked into whether NVP-231 influences cancer cell responses and can reduce CerK activity in intact cancer cells. In order to do this, a human CerK-containing cDNA construct was transfected into the MCF-7 breast cancer cell line in a stable manner. Following this, the cells were treated with NBD-ceramide, a cell permeable fluorescently labeled analogue of C6-ceramide that functioned as a CerK substrate to undergo phosphorylation. Cellular CerK activity, as indicated by NBD-C1P production, was gradually decreased when cells were treated with increasing doses of NVP-231, suggesting that in transfected cells, NVP-231 is active. In the cellular system, the IC50 for CerK was determined to be 59.70 ± 12 nM. Additionally, we evaluated NVP-995, an inactive molecule with the identical chemical structure but two extra methoxy groups [11]. By using the cellular CerK activity assay, we were able to demonstrate that NVP-995 exhibited no inhibitory effects In addition, we measured the amounts of C16-C1P and C16-ceramide in MCF-7 cells treated with NVP-231. Following NVP-231 treatment, C16-C1P decreased concentration-dependently, whereas total ceramide levels rose. The analysis of the several ceramide subspecies revealed that C16-, C24-, and C24:1-ceramides were present in the largest amounts. Ceramides from the subspecies C18, C18:1-, C20, and C22 were hardly noticeable (Figure 1).

Figure 1.Effect of NVP-231 and NVP-995 on CerK activity in transfected MCF-7 cells. A) MCF-7 cells that were either left untransfected (white column, −) or transfected with a cDNA of human CerK (hCerK, +) were treated for 2 h with the indicated concentrations of NVP-231 (grey columns) or NVP-995 (black column) in the presence of NBD-C6-ceramide as described in the Methods section. Cells were harvested and taken for lipid extraction. Lipids were separated by TLC and analysed on a fluorescence imaging system. The density of spots corresponding to phosphorylated NBD-ceramide was measured and results expressed as % of CerK activity compared with Wt control cells. Data are means ± SD (n=4). (B and C) MCF-7 cells were treated for 24 h with the indicated concentrations of NVP-231. B) Lipids were then extracted and taken for LC-MS/MS to quantify C16-C1P and C) the various ceramide subspecies. Results are expressed as pmol lipids per 106 cells and are means ± SD (n=3). *P<0.05, **P<0.01, ***P<0.001 considered statistically significant when compared with the control samples; ###P<0.001 statistically significant when compared with the hCerK overexpressed
untreated samples.
One of the physiological roles of C1P that has been documented in the literature is promoting cell division [12,13], albeit the specific processes involved are yet unknown. Furthermore, increased CerK expression in breast cancer patients is associated with a worse prognosis, even indicating CerK as a prognostic marker in breast cancer, according to Ruckhäberle E.
Here, we looked at whether cellular CerK contributes to the survival and growth of cancer cells and whether CerK inhibition affects the behavior of cancer cells. Viability was concentration-dependently decreased after 48 hours of NVP-231 treatment for either MCF-7 or NCI-H358 cells, with an IC50 of roughly 1 μM for MCF-7 cells and 500 nM for NCI-H358 cells (Figure 2).

Figure 2. Effect of NVP-231 and NVP-995 on the viability of MCF-7 and NCI H358 cells. A and B) MCF-7 (A) and NCI-H358 (B) cells were plated in a 96-well black plate (1 × 104 cells per well) and treated for 48 h with either vehicle (Co) or the indicated concentrations of NVP-231 (in nM). For the last 4 h of treatment, alamarBlueR was added and fluorescence was determined as described in the Methods section. Data are expressed as % of control and are means ± SD (n=4). C and D) MCF-7 (C) and NCI-H358 cells (D) were plated in a 96-well plate at a density of 1 × 104 cells per well and treated with the indicated concentrations NVP-231 or NVP-995 for 72 h. For the last 24 h, BrdU was added to the culture medium. Incorporated BrdU was measured by elisa using an anti-BrdU antibody according to the manufacturers' protocol. Data are expressed as % of BrdU incorporation compared with the control group and are means ± SD (n=4). E and F) MCF-7 cells (E) and NCI-H358 cells (F) were treated with the indicated concentrations of NVP-231 and NVP-995 in growth medium and incubated for further 10 days (NCI-H358 cells) or 14 days (MCF-7) to allow colony formation. Cells were stained with 2% (w·v−1) crystal violet and the numbers of colonies containing more than 50 cells were counted. Data are expressed as % of control and are means ± SD (n=4). *P<0.05, **P<0.01, ***P<0.001 considered statistically significant when compared with the control groups.
By observing the incorporation of BrdU into newly produced DNA, we were able to quantify the impact of NVP-231 on DNA synthesis even further. After 72 hours, both cell lines' DNA production was decreased by NVP-231 treatment. In both cell lines, a 60–70% reduction was seen after 72 hours with 1 μM of NVP-231, the highest concentration tested. Furthermore, a clonogenic test was used to track the MCF-7 and NCI-H358 cells' ability to form colonies after they were treated with NVP-231 for ten to fourteen days. Following NVP- 231 treatment, both cell lines displayed decreased colony formation; complete suppression was achieved with 500 nM in NCI-H358 and 1 μM in M phase cell. In order to learn more about the cause of the decreased viability and DNA synthesis of we examined the quantity of PI uptake in both cell lines after they were treated with NVP-231 as a gauge of cell death. After being treated for 72 hours, the latest time point examined, the number of PI-positive dead cells grew steadily with treatment with 1 μM NVP-231, reaching 20% in MCF-7 cells and more than 40% in NCI-H358 cells (Figure 3).

Figure 3. Effect of NVP-231 on cell death of MCF-7 and NCI-H358 cells. MCF-7 (A and C) and NCI-H358 (B and D) cells were treated for 24–72 h with either vehicle (Co, 0) or 1 μM NVP-231. Cells were collected and stained with presidium iodide and analysed by flow cytometry to detect PI-positive cells (A and B). Data are expressed as % of cells with positive PI staining and are means ± SD (n=3). *P<0.05, **P<0.01, ***P<0.001 considered statistically significant when compared with the control groups. In parallel, cell lysates were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis (C and D) using antibodies against cleaved caspase-3 (upper panels), cleaved caspase-9 (middle panels) and GAPDH (lower panels) at dilutions of 1:1000 each. Data show duplicate samples and are representative of three independent experiments giving similar results. (E and F) Graphs show the densitometry evaluation of Western blot data of MCF-7 (E) and NCI-H358 cells (F). Results are expressed as % of control and are means SD (n=3). *P<0.05, **P<0.01, ***P<0.001 considered statistically significant when compared with the control groups.
We examined the cell lysates for indicators of apoptosis, such as cleaved caspase-3 and caspase-9, to determine whether the enhanced cell death was caused by programmed cell death. Notably, it was previously shown that MCF-7 cells lack caspase 3. Shows, the MCF-7 cell line employed in this investigation expresses caspase-3 unambiguously. NVP-231 induced increased caspase-3 and caspase-9 cleavage in both cell lines. The two cell lines' temporal courses, however, differed. The maximum caspase-3 and caspase-9 cleavage and consequent activation in MCF-7 cells happened at 24 hours, and then it declined once more, indicating that necrosis-like alterations happened later on, which explains the high number of 48 and 72 hours' worth of PI-positive cells. Cleavage of caspase-3 and caspase-9 continued after 72 hours in NCI-H358 cells.
M phase arrest results with NVP-231 therapy
We also looked into how NVP-231 affected the development of the cell cycle. Notably, there was a difference between the two cell lines' regular constitutive distribution of growing cells during the cell cycle. Compared to NCI-H358 cells, MCF-7 cells displayed a notably higher proportion of polyploidy cells (>4N DNA content), but less cells in the S phase and sub-G1 phase—which includes cells with fragmented DNA typical of apoptotic/necrotic cells. After being treated with increasing doses of NVP-231 for 24 hours, either MCF-7 (Figure 4A and C upper panels) or NCI-H358 (Figure 4B and C lower panels) cells showed a decrease in the number of S phase cells and an accumulation of G2/M (4N DNA content) cells. Meanwhile, more cells carrying broken within the sub-G1 fraction, DNA was found. NVP-231 had no effect on the proportion of polyploidy cells (>4N DNA content) in either of the two cell lines. As anticipated, the distribution of cell cycles was unaffected by the inert substance NVP-995 (Figure 4).

Figure 4. Effect of NVP-231 and NVP-995 on cell cycle progression of MCF-7 and NCI-H358 cells. MCF-7 (A) and NCI-H358 (B) cells were treated for 24 h with either vehicle (Co) or the indicated concentrations of NVP-231 and NVP-995 (in nM). Cells were collected, stained with propidium iodide and analysed by flow cytometry for DNA content. Data are expressed as % of cells in the corresponding cell cycle phases and
are means ± SD (n=6). *P<0.05, **P<0.01 considered statistically significant when compared with the respective control groups. (C) Graphs show representative samples of MCF-7 control cells (upper left panel), MCF-7 cells treated with 1000 nM NVP-231 (upper right panel), NCI-H358 control cells (lower left panel) and NCI-H358 cells treated with 1000 nM NVP-231 (lower right panel).
In order to examine the reported G2/M cell accumulation more thoroughly, we looked at whether cell arrest occurred during the M phase or at the G2/M boundary as a result of CerK inhibition. In order to do this, phosphorylated histone H3, a recognized marker for mitosis, was examined [14]. When NVP- 231 was administered to MCF-7 cells (Figure 5A) and NCI-H358 cells (Figure 5B), flow cytometry revealed a concentration-dependent rise in the cells' mitotic index. Simultaneously, phospho-Ser10 histone H3 in cellular lysates was analyzed using Western blot, and the results indicated that NVP-231 treatment also improved staining (data not shown). These findings show that cells treated with NVP-231 underwent M phase arrest as opposed to G2/M boundary arrest (Figure 5).

Figure 5. Effect of NVP-231 on the mitotic index of MCF-7 and NCI-H358 cells. MCF-7 (A) and NCI-H358 (B) cells were treated for 24 h with either vehicle (−) or the indicated concentrations of NVP-231 (in nM). Cells were collected and stained withan anti-phospho-Ser10 histone H3 antibody and analysed by flow cytometry. Data are expressed as % mitotic cells and are means ± SD (n=6). *P<0.05, ***P<0.001 considered statistically significant when compared with the control groups.
Next, we looked into other chemical elements that are known to play a role in the regulation of the cell cycle, particularly in the G2/M transition. The so-called mitosis-promoting factor, a heterodimeric protein made up of cyclin B and cdc2 (CDK1), is known to influence this transition. NVP-231 treatment resulted in a concentration-dependent decrease in CDK1 phosphorylation at Tyr15 and an up-regulation of cyclin B1 phosphorylation at Ser133 in the two cell lines under study. The discovery that cells did not get stopped in G2, but instead entered the M phase, is further supported by the need for both events during the G2/M transition. On the other hand, when CerK was inhibited, total CDK1 expression similarly decreased Additionally, we discovered that wee1, a kinase that controls CDK1 activity, was down-regulated by NVP-231 Furthermore, we found that CDK4, a kinase crucial for the G1/S transition, was downregulated (Sheppard and McArthur, 2013). Over the course of 72 hours, there was a time-dependent decrease in CDK4 protein expression, which was more noticeable in NCI-H358 cells than in MCF-7 cells this difference in expression may be due to the shorter doubling time of NCI-H358 cells compared to MCF-7 cells. The decreased number of cells in S phase could be explained by this down-regulation of CDK4 (Figures 6 and 7).

Figure 6. Effect of NVP-231 and NVP-995 on cyclin B1, CDK1 and wee1 expressions in MCF-7 and NCI-H358 cells. MCF-7 (A) and NCI-H358 (B) cells were treated for 24 h with either vehicle (Co) or the indicated concentrations of the NVP-231 and NVP-995 (in nM). Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against phospho-Ser133 cyclin B1, total cyclin B1, phospho-Tyr15 CDK1, total CDK1, wee1 and GAPDH. Data show duplicate samples and are representative of at least three independent experiments giving similar results. (C and D) Graphs show the densitometric evaluation of Western blot data of MCF-7 (C) and NCI-H358 cells (D). Results are expressed as % of control and are means SD (n=4–6). *P<0.05, **P<0.01, ***P<0.001 considered statistically significant when compared with the control groups.

Figure 7. Time-dependent effect of NVP-231 on CDK4 protein expression in MCF-7 and NCI-H358 cells. MCF-7 (A) and NCI-H358 (B) cells were treated for 24 h with either vehicle (Co) or for the indicated time periods with 1 μM of NVP-231. Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against CDK4 and β-actin. Data show duplicate samples and are representative of three independent experiments giving similar results. (C) Graphs show the densitometric evaluation of Western blot data of MCF-7 and NCI-H358 cells. Results are expressed as % of control and are means ± SD (n=6). **P<0.01, ***P<0.001 considered statistically significant when compared with the control groups. ###P<0.001 compared with the NCI-H358 control group.
Drug-induced apoptosis sensitizes cells when CerK is inhibited
NVP-231 and staurosporine were given to the cells concurrently to examine if CerK inhibition makes cancer cells more susceptible to druginduced apoptosis. In order to more accurately evaluate the impact of NVP- 231, staurosporine—a non-specific yet powerful inducer of apoptosis—was employed at a low dose. When compared to control cells, NCI-H358 cells treated with 20 nM staurosporine displayed a twofold increase in DNA fragmented cells (Figure 8A). Staurosporine induced a synergistic rise in DNA fragmentation when cells were pre-treated with increasing amounts of NVP-231 It's interesting to note that MCF-7 cells responded to staurosporine differently than NCI-H358 cells. There was only a slight increase in DNA fragmentation when using the same concentration of 20 nM staurosporine (data not shown). Consequently, we took measurements staurosporine-induced G2/M arrest and discovered that NVP-231 pretreatment of cells did, in fact, increase the G2/M arrest caused by staurosporine Next; we employed RNA interference to downregulate the enzyme in order to prevent CerK from acting. Staurosporineinduced DNA fragmentation was considerably up-regulated in NCI-H358 and MCF-7 cells when CerK mRNA expression was lowered to 30–40% of control transfected cells (Figure 8).

Figure 8. Effect of NVP-231 and CerK down-regulation with siRNA on staurosporineinduced cell death. NCI-H358 (A) and MCF-7 cells (B) were pretreated for 4 h with either vehicle (DMSO, 0) or the indicated concentrations of the NVP-231 and NVP-995 (in nM) before the addition of 20 nM staurosporine (+) for an additional 20 h. Cells were collected and analysed for DNA fragmentation and G2/M arrest. Data are means ± SD (n=6). *P<0.05, **P<0.01, ***P<0.001 considered statistically significant when compared with the untreated control group; ###P<0.001 compared with the staurosporine-treated control group; §§§P<0.001 compared with the corresponding NVP-231-treated groups. NCI-H358 (C) and MCF-7 cells (D) were transfected with a control siRNA (ctrl-siRNA) or siRNA of CerK (siCerK) and then treated for 20 h with the indicated concentrations of staurosporine (in nM). Cells were collected and analysed for DNA fragmentation. Data are means ± SD (n=3). *P<0.05, ***P<0.001 considered statistically significant when compared with the untreated control group; ##P<0.01, ###P<0.001 compared with the untreated siCerK group; §P<0.05, §§P<0.01, §§§P<0.001 compared with the corresponding staurosporine-treated groups.
Finally, we tested whether overexpression of CerK in MCF-7 cells protects from drug-induced apoptosis. To this end, cells were transiently transfected with a human CerK cDNA construct prior to treatment with staurosporine. As shown in Figure 9A, staurosporine reduced MCF-7 cell viability in a concentrationdependent manner. When CerK was overexpressed, the proapoptotic effect of staurosporine was reversed, suggesting that endogenously produced C1P may promote cell viability. Moreover, overexpression of CerK reduced the NVP-231-triggered increase of phospho-histone H3 (Figure 9).

Figure 9. Effect of CerK overexpression on staurosporine-stimulated apoptosis and NVP-231-mediated histone H3 phosphorylation. A) Cells transfected with the empty vector (pcDNA3) or with a cDNA construct of hCerK (pcDNA3-hCerK) were plated in a 96-well black plate (1 × 104 cells per well) and treated for 48 h with either vehicle (Co) or the indicated concentrations of staurosporine (in nM). For the last 4 h, alamarBlueR was added and fluorescence was determined as described in the Methods section. Data are expressed as % of control and are means ± SD (n=5), **P<0.01, ***P<0.001 considered statistically significant when compared with the corresponding value of pcDNA3 transfected samples. B) NCI-H358 cells transfected with the empty vector (pcDNA3) or with a cDNA construct of hCerK (pcDNA3-hCerK) were treated for 24 h
with either vehicle (Co) or the indicated concentrations of NVP-231. Cells were then taken for phospho-histone H3 analysis by flow cytometry. Results are expressed as % of phospho-H3 positive cells and are means ± SD (n=3). ***P<0.001 when compared with the pcDNA3 transfected control samples; §P<0.05, §§§P<0.001 when compared with the corresponding NVP-231-treated pcDNA3 transfected samples.
Conversation
Prior research indicated that CerK and its by product C1P regulated both apoptosis and cell growth. The precise mechanics, meanwhile, are still mostly unknown. It was initially demonstrated that CerK plays a significant role in regulating plant cell death. Plants that were mutated for the plant CerK homologue accelerated cell death 5 (acd5) in Arabidopsis exhibited both increased cell death following pathogen infection and an accumulation of ceramides. Subsequently, down-regulating CerK by siRNA transfection led to decreased DNA synthesis and enhanced death in an in vitro system containing human lung cancer cells. Numerous investigations used exogenous C1P injected to cells to reveal the lipid C1P's cellular activity. Cultures either diluted in solvents containing dodecanal or at quite high concentrations as liposomes or micelles. By employing these methods, it appears that C1P causes either proliferation or death, depending on the kind of cell [15]. Recently, Gangoiti P, demonstrated that exogenous C1P stimulated glycogen synthase kinase- 3β phosphorylation, increased retinoblastoma (Rb) phosphorylation, and enhanced cyclin D1 expression in C2C12 myoblasts, therefore inducing cell proliferation. It is evident that C1P is a cell-impermeable lipid that requires a transporter to enter the cell or, in the case of dodecanal, pore creation to occur.
As an alternative, exogenous C1P may function through a cell surface receptor, just as S1P. But as of now, neither a dedicated C1P export mechanism, such as a transporter, nor a There is a known, highly-affinity cell surface receptor for C1P. Thus, it is still unclear whether the cellular effects of extracellular C1P are indeed mediated by a particular action on a putative receptor or stem, at least partially, from membrane disruptions such as those brought on by detergents. However, as C1P is mostly produced inside of cells, its preferred site of action is probably intracellular. In the current investigation, we employed NVP-231, a recently created strong catalytic inhibitor of CerK that suppresses CerK in the low nM range in both transfected cells and cell-free in vitro environments (Figure 1). We demonstrate that in two different histo types of cancer cell lines, namely the MCF-7 breast cancer cell line and Treatment with NVP-231 concentration-dependently decreased colony formation, DNA synthesis, and cell survival in the lung cancer cell line NCI-H358. The cells' aberrant cell cycle progression resulted in a notable build-up of M phase cells and, in addition, decreases in S phase cell count. Later on, the rate of cell death rose. Without a doubt, an inhibitor's selectivity is important in pharmacology. Given that NVP- 231 was discovered to be a ceramide-competitive CerK inhibitor, it's probable that the inhibitor impedes other enzymes that have a lipid affinity, specifically ceramide, if it does so at all. Graf C, addressed the specificity of NVP-231 for CerK (2008a). We also evaluated other lipid kinases and other ceramideaffinity- displaying enzymes. The most susceptible enzyme to NVP-231 was DAGKα, which had an IC50 of 5 μM, inhibiting it, while the IC50 values of the other enzymes tested ranged from 10 to 100 μM. The expression and activity of four CDKs, including CDK1 (Cdc2), CDK2, CDK4, and CDK6, as well as their regulatory cyclin partners, which include cyclin A, B, D, and E, are typically linked to the regulation of cell cycle progression in mammalian cells. A kinase that is vital to cell cycle progression, cell division, and viability, CDK1 malfunction is lethal. According to Enserink and Kolodner, CDK1 is closely related to DNA replication, chromosomal cohesion and condensation, spindle assembly, location, and stability, as well as chromosome attachment, alignment, and segregation. Each of these occurrences could be interfered with when CDK1 function is impaired. Similar to our work, Que XY, recently showed that silencing CDK1 in osteosarcoma cells led to decreased cell proliferation and accumulation in G2/M as well as a decrease in the number of cells in S phase. Furthermore, Rani CS, demonstrated that treating fibroblasts with the glucosylceramide synthase inhibitor 1-Phenyl-2-Decanoylamino-3- Morpholino-1-Propanol (PDMP) resulted in an increase in cellular ceramide and a decrease in glucosylceramide in addition to inducing a reduction in S phase and an accumulation of cells in G2/M. They also noticed that when CDK1 was treated with PDMP, its activity decreased. The authors proposed that ceramide in particular was accountable for G2/M arrest, whereas the reduction in S-phase cells was attributed to the loss of glucosylceramide. Furthermore, Drobnik W, demonstrated that cellular ceramide concentrations were elevated in fibroblasts from Tangier disease patients, which are distinguished by a disrupted lipid metabolism because of a mutated ABC1 transporter. In response to various mitogens, cells accumulated in G2/M and vanished from S phase. Given that ceramide accumulates when CerK is inhibited or knocked out, it is plausible that various treatment approaches stimulate related pathways that result in the same cellular phenotype. However, due to conflicting information regarding ceramide's function in cell cycle regulation, it is still unclear if this phenotype is mediated by ceramide. Ceramide was further demonstrated to arrest cells in G1, including MCF-7 cells. This process appears to entail Rb protein dephosphorylation and increased expression of the CDK inhibitory factor p21. In a different study, it was demonstrated that myriocin inhibits the synthesis of new sphingolipids, leading to a decrease in ceramide and other sphingolipids, a reduction in DNA synthesis and melanoma cell proliferation, and an accumulation of cells in G2/M phase that eventually disappears from S phase. The addition of exogenous C8-ceramide entirely prevented the considerable loss of CDK1 protein that myriocin was demonstrated to generate on a molecular level. This led the investigators to believe that ceramide or one of According to Lee, its catabolites are crucial for CDK1 expression. As ceramide-converting enzymes like ceramidases and sphingosine kinases are known to be important regulators of ceramide's actions, the ceramide effect undoubtedly varies depending on the type of cell in question and is dependent on the equipment within the cell [1]. Upon attempting to investigate the signal's upstream, we discovered that NVP-231 inhibited wee1 expression in both NCI-H358 and MCF-7 cells. It has been revealed that wee1 must be degraded by an E3 ubiquitin ligase activity to ensure entry into mitosis. Wee1 kinase is a crucial regulator of CDK1 activity by regulating Tyr15 phosphorylation and consequently CDK1 inactivation [16]. Thus, it is easy to assume that CerK Increased E3 ubiquitin protein ligase activity as a result of inhibition encourages wee1 degradation and the start of mitosis.
Our findings further show that CDK4 protein was down-regulated in a concentration- and time-dependent manner when NVP-231 was administered to cancer cells. In synchronized cells, this effect was even more noticeable (data not shown). According to Sheppard and McArthur, CDK4 is a key component of the p16/cyclin D/CDK4 and 6/retinoblastoma pathway, which promotes the G1 to S cell cycle transition. Moreover, CDK4 inhibition or downregulation enhances cell death in addition to causing G1 arrest. The observed decrease in cells from S phase and higher apoptosis could be explained by the observed down-regulation of CDK4 caused by CerK inhibition. However, more research is needed to fully understand the mechanism underlying these benefits are required to deal with this problem. The question of which lipid subspecies—the increased ceramide, the lost C1P, or some other metabolite— is actually causing the consequences is particularly significant. In this context, it's also important to note that intestinal polyps from SphK1-deficient animals had down-regulated CDK4 expression, which was associated with decreased epithelial cell proliferation. Exogenous sphingosine, but not ceramide, could replicate the effect of decreased CDK4 expression and proliferation in that studies in vitro studies using rat intestinal epithelial cells. Conversely, Kim WH, demonstrated that ceramide lowered CDK4 activity in HL-60 cells, which led to G1 arrest and eventual apoptosis. Therefore, more research is required to elucidate how the various sphingolipid species function within the cell cycle machinery. Not only can cell cycle-regulating activities take place in the nucleus, but they can also happen in the cytoplasm. Given this, figuring out CerK's subcellular location within the dynamic range of the cell cycle will be essential to determining potential C1P direct targets. Although it hasn't been confirmed, a nuclear role is highly expected because CerK's sequence includes nuclear import and export signals. The location of wild-type CerK in the Golgi, cytoplasm, and nucleus was shown by Rovina P, using COS-1 cells transfected with a GFP-coupled CerK construct. Given these findings, it is intriguing to hypothesize that CerK plays a role in the center. Additionally, Chen WQ, previously demonstrated that CerK has phosphorylation sites other than Ser408, such as Ser340. The conventional phosphorylation sites for proline-directed PKs, such as CDKs and MAPKs, are represented by these two residues. It is quite likely that CerK is coupled to the cell cycle in one way or another because both of these families of PKs play critical roles in regulating the cell cycle and promoting cell proliferation.
Making cancer cells more susceptible to proapoptotic chemotherapy is a fundamental component of effective anti-cancer treatment. According to our research, CerK inhibition alone is adequate to cause cancer cells to die. Moreover, it makes the cells more susceptible to drugs that cause death, including the unspecific kinase inhibitor staurosporine. CerK inhibition, not an unidentified source, is responsible for this sensitizing impact. Impact, since cells that overexpressed CerK were less vulnerable to the death-inducing effects of staurosporine. Gomez-Muñoz A, also suggested that C1P plays a part in cell survival. These scientists demonstrated that C1P increased cell survival in exogenous murine macrophages by upregulating the anti-apoptotic protein Bcl-xL and activating PI3K and Akt/PKB. Nevertheless, in our cell systems, overexpressing CerK did not result in increased Akt (PKB) or p42/ p44-MAPK phosphorylation (data not shown). This rule outs C1P's involvement in early survival signaling and lends credence to our theory that C1P promotes M phase transition by acting inside the nucleus. Additional research is required to determine the most promising combinations of NVP-231 with traditional anticancer drugs to maximize the synergistic anti-cancer effect not only in in vivo in patients, but ultimately also in vitro in cancer cell lines. Our observations also point to a possible therapeutic application of NVP-231 in other proliferationassociated disorders, as CerK plays a critical role in normal mitotic transition and cell proliferation. Therefore, the use of NVP-231 to treat mesangioproliferative glomerulonephritis is an appealing novel method. In this line, we have recently demonstrated that in renal mesangial cells, CerK deletion or NVP-231 therapy inhibits cell proliferation. Nevertheless, up to now, only breast cancer has been linked to increased CerK expression—other illnesses are not. Consequently, Ruckhäberle E, found a correlation between increased CerK expression and a poorer prognosis for individuals with breast cancer, even speculating that CerK might be a beneficial breast cancer prognostic indicator.
To summarize, our research reveals CerK's critical function in M-phase control for the first time and suggests that proapoptotic anti-cancer drugs and targeted CerK inhibition could be combined for more potent cancer treatment.
None.
None.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
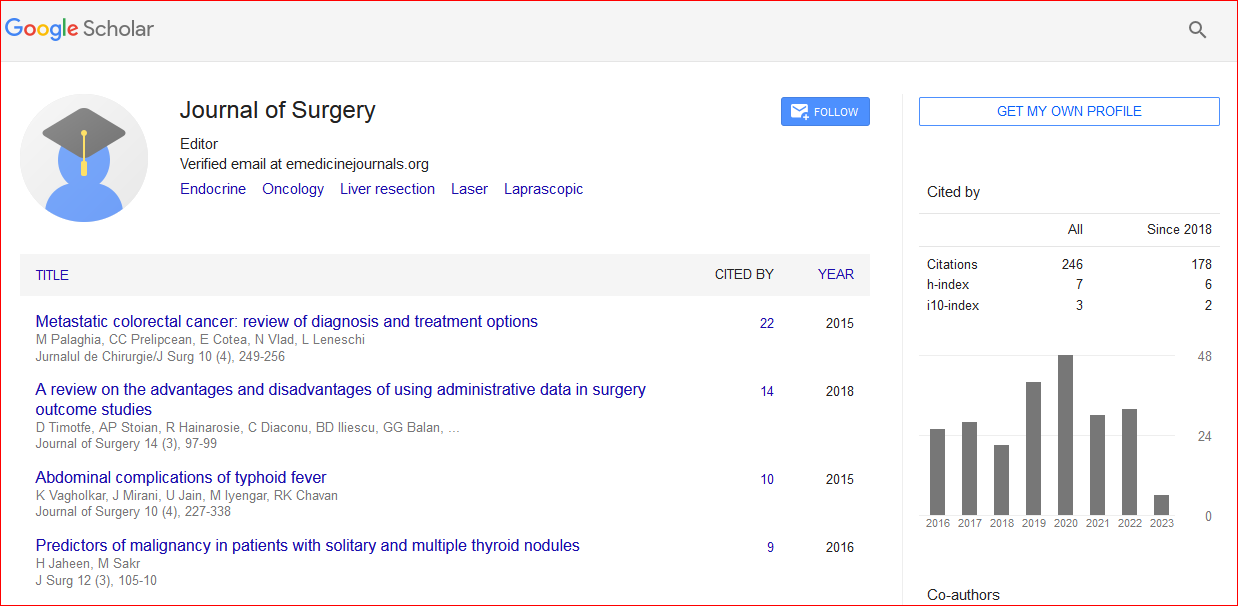
Journal of Surgery received 288 citations as per Google Scholar report