Research - (2022) Volume 10, Issue 3
Received: 02-Mar-2022, Manuscript No. jnd-22-56780;
Editor assigned: 04-Mar-2022, Pre QC No. P-56780 PQ);
Reviewed: 18-Mar-2022, QC No. Q-56780;
Revised: 23-Mar-2022, Manuscript No. R-56780;
Published:
30-Mar-2022
, DOI: 10.4172/2329-6895.10.3.482
Citation: Akram El-Sayed, Al-Baraa “Quality Control of Mitochondria in Parkinson's Disease (PD) Using ATP and Bradford Assays” J Neurol Disord 9(2022):482.
Copyright: © 2022 Sayed El-Akram. This is an open-access article distributed under the terms of the creative commons attribution license which permits
unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited
Parkinson's disease is a heterogeneous, multifactorial and often complex disease characterized by motor impairment due to the presence of Lewy bodies and prominent degeneration of dopaminergic neurons in the substantia nigra. Although the specific pathogenesis involving PD remains under investigation, mitochondrial dysfunction has been widely accepted as one of the major pathogenic pathways underlying the development of PD. Based on the hypothesis that depiction of HtrA2 (serine protease gene, mitochondrial precursor) might contribute to an increase in mitochondrial stress and transcriptional up regulation of the nuclear stress response CHOP gene. The present study aimed to analyze through laboratory based research the role of HtrA2 and CHOP in the transmission of stress signaling and the consequent activation of mitochondrial quality control in Parkinson's disease using ATP and Bradford assays.
Parkinson's disease • Lewy bodies • Mitochondrial dysfunction • Bradford assays
Definition
Parkinsonism is a progressive neurological disorder of muscle movement characterized by any combination of tremor, rigidity, bradykinesia and progressive postural instability. Cognitive impairment is sometimes prominent.
Causes
The cause of the disease is unknown for most patients. The disease (especially idiopathic type) may be due to: Destruction of dopaminergic neurons in the substantia nigra which is a part of the extrapyramidal system with a consequent reduction in dopamine action in the corpus striatum of the basal ganglia responsible for motor control and overproduction and/or overactivity of acetylcholine triggers abnormal signaling, leading to loss of control of muscle movements.
Drug therapy of Parkinsonism
The central imbalance between the dopaminergic system (reduced dopamine activity) and cholinergic system (increased acetylcholine activity) is the backbone of designing drug therapy for the disease.
Amantadine
It is an influenza antiviral agent; it has an indefinite mechanism as an antiparkinsonian drug. Its mechanism of action is not definitely known, but it may work by increase dopamine release, inhibit cholinergic system, blockade of NMDA excitatory receptors.
It may be used in mild cases of the disease. It is less effective than levodopa but more effective than anticholinergic drugs. Tolerance develops to its effect after 6-8 months of continued treatment. Its side effects include anorexia, nausea, constipation, postural hypotension, restlessness and confusion.
Mitochondrial structure and function
As shown in Figure 1, mitochondria are highly regulated membrane bound organelles involved in fundamental cellular and metabolic functions within eukaryotic cells, such as energy supply (ATP production) through oxidative phosphorylation (OXPHOS), calcium and metabolite supply, inflammation, intracellular signaling, cell proliferation and apoptosis [1].
Figure 1. Illustration of the structure of mitochondria showing the outer and inner membranes, matrix, cristae, mitochondrial DNA and ribosomes.
The function and survival of dopaminergic neurons strongly rely on appropriate mitochondrial homeostasis. Mitochondrial dysfunction has been widely accepted as one of the major pathogenic pathways underlying the development of PD [2].
Parkinson's disease (PD) is the second most prevalent age related neurodegenerative disorder of the central nervous system (CNS), affecting over 2% of the worldwide population older than 65 years.
The neuropathology of PD is characterized by the presence of Lewy bodies and prominent degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), which leads to severe striatal dopamine (DA) deficits, resulting in motor impairment. Bradykinesia, resting tremors, slowness of movement, muscular rigidity and postural instability comprise some of the clinical motor features of PD. Patients may also develop nonclinical manifestations such as autonomic impairment, cognitive variations, depression, sleep disturbances and late stage dementia.
Changes in mitochondrial structure and dynamics, dysregulation of transcription factors, impairment and aggregation of protein components due to nuclear DNA and mitochondrial DNA (mtDNA) gene mutations or environmental stress are important causes associated with mitochondrial dysfunction [3]. Accordingly, mutations in a broad range of primary genes have been implicated in mitochondrial dysfunction and are known to play a role in autosomal dominant and recessive forms of PD, such as u-Synuclein (SNCA, PARK1), Parkin (PARK2), Phosphatase and tensin homolog (PTEN)-induced putative kinase I (PINK/, PARK6), Leucine-rich repeat kinase 2 (LRRK2) and DJ-/(PARK7) [4].
Dysfunctions at mitochondria are removed via lysosomal degradation through the process of organellar quality control, so; called mitophagy as shown in Figure 2. Mitophagy is triggered by numerous cellular events, including cellular stress and differentiation [5]. This process is done by complexes of molecules that recognize dysfunction at mitochondria and target them for degradation through the autophagy or/mitophagy process done by ATG complexes importantly two of the genes that are mutated in PD, PINK/and Parkin are coordinators of the PINK I/Parkin-mediated mitophagy process [6].
Figure 2. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemiaــreperfusion injury-science direct.
Mice without function at HtrA2/0 mi protease activity showed induced up regulation of CHOP, reduced mitochondrial function and premature aging in brain cells, due to an increase in mtDNA deletions [7].
ATP assay
ATP levels were assessed using Cell Titer-Glo (Promega) following the manufacturer's instructions. Briefly, the genotypes were cultured in a 96 well plate, followed by drug treatment. For analysis, ATP levels were quantified using ATP standard curves using serial dilutions of ATP (50 μL) solution in culture medium. Cells were incubated for 30 minutes. Chemiluminescence as shown in Figure 3 was measured using a SpectraMax (MS).
Figure 3. ATP assay illustrating the transformation of luciferin into oxyluciferin as an example of a chemiluminescence reaction.
Bradford assay
Protein levels were determined for normalization purposes using the Bradford assay. Briefly, the cells cultured in 96 well plates, as shown in Figure 4, were washed with phosphate buffered saline (PBS) and incubated with Bradford solution (Sigma, Germany) for 30 minutes, followed by subsequent washing in PBS. In each experiment, samples were normalized to a standard curve prepared by a BSA stand and stock solution (in PBS) with serial dilutions made in the Bradford solution.
Figure 4. 96-well plates of Bradford assay.
Statistical analysis
Data were collected from biologics in triplicate for each genotype. All results are presented as the mean ± SEM and were analyzed using one way or two ways ANOVA.
The different genotypes analyzed in this research were treated with 50 μM of the neurotoxin 6-OHDA and 0.5 μM of ADEP4 and ACP5. Then the cell viability was determined by MTT Assay. Cell viability showed a concentration dependent reduction in the viability level of the different genotypes (Figures 5A and 5B). Compared to the vehicle control (DMSO), cell viability was mainly reduced in the HtrA2 KO/CHOP genotype. Depletion of HtrA2 significantly increased the susceptibility to treatment with 6-OHDA (p<0.0001).
Figure 5. CHOP loss of function impairs transcriptional activation of mitochondrial quality control signaling. MEFs from wild-type (WT) (orange columns), CHOP-knockout (KO) (purple columns) and HtrA2 KO mice (green columns) were treated with ADEP4 and ACP5. RTـــPCR analysis illustrated that depletion of HtrA2 results in significant up regulation of the molecular quality control marker (A) Hsp60 and organellar quality control marker (B) The data is represented as the mean ± SEM. The statistical comparison was performed by two-way ANOVA followed by Tukey's multiple comparisons versus the vehicle control and between the indicated groups with P <0.05. *P<0.05, **P<0.01 ***P<0.001.
These results display a drug concentration dependent effect on the cell viability of the genotypes, indicating that depletion of cellular content might be due to high concentrations of drug treatment with 6-OHDA. Therefore, the concentration of the drug will be proportional to the number of living cells as shown in Figure 6.
Figure 6. ATP production is decreased in the HtrA2 KO/CHOP genotype. Depletion of HtrA2 significantly increased the susceptibility to drug treatment Compared to vehicle control (DMSO), loss of energy production was significantly higher in HtrA2 KO/CHOP genotype treated only with 6-OHDA. The energy level was significantly decreased in the HtrA2/CHOP KO genotype treated in conjunction with ADEP4 (A) or ACP5 (B). ATP product ions were significantly higher in the HtrA2/CHOP KO genotype (the data are presented as the mean ± SEM). The statistical comparison was performed by one or two-way ANOVA followed by Bonferroni's multiple comparisons versus the vehicle control and between the indicated groups with P < 0.05. *P<0.05, **P<0.01, ***P<0.001.
ATP production is decreased in HtrA2 KO/CHOP genotype
ATP production was determined via ATP assay nominalized by the measurement of protein levels using the Bradford method. Likewise, in the cell viability assay, the ATP assay also displayed a concentration dependent reduction in energy production among the different genotypes and reflected both the cell viability and the metabolic status of the cells. The results illustrated that depletion of HtrA2 significantly increased susceptibility to drug treatment as shown in (Figure 7). Compared to the vehicle control (DMSO), the loss of energy production was significantly higher in the HtrA2 KO/CHOP genotype treated only with 6-OHD A (p<0.0001).
Figure 7. Microscopy visualization of mouse embryonic fibroblasts (MEFs) obtained from wild-type (WT), CHOP knockout (KO) and HtrA2 KO mice treated with 6-OHDA and ACP5. Compared to the other treatments, the HtrA2/CHOP KO genotype had a significantly lower concentration of live cells than the well treated with only ACP5, which significantly improved cell survival.
The HtrA2 gene comprises 4152 bases of DNA located in the forward strand of chromosome 2 and composed of eight exons. The gene encodes a 49 kDa protein of 458 amino acid (aa) residues. The full length translated protein is located within the mitochondrial intermembrane space (IMS) and contains a trimeric pyramidal structure that goes through a multifaceted allosteric activation pathway and defines HtrA2 proteolytic regulation, specificity and activity. The protease comprises the Nterminal mitochondrial localization sequence (1-40 aa), a transmembrane domain (105-125 aa) that dictates the topology of the protein, a serine protease domain (150-343 aa) with catalytic triad residues (Ser306, His198 and Asp228), and one PDZ domain (364-445 aa) located at the C-terminal end [8].
HtrA2 proteases are able to activate or coordinate heterogeneous signaling pathways implicated in PQC. Following apoptotic stresses, the N-terminus is cleaved, and the mature HtrA2 protein is released from the mitochondrial intermembrane space (IMS) into the cytosol. Numerous structural studies have presented an overall analysis of the molecular architecture of human HtrA2, although the accurate pathway by which the protein is activated remains unclear [9].
In mitochondrial biogenesis, HtrA2 predominantly operates as a chaperone, where it prevents polypeptide aggregation and assists with correct protein folding. Although movement of the PDZ domain, facilitated by stress conditions such as temperature increase or by PDZ binding peptides, was suggested to trigger HtrA2 activity, which initiates its proteolytic function and degrades the impaired proteins, similar to the allosteric mechanism suggested for the regulation of its bacterial homolog (HtrA/DegP).
Genomic studies have demonstrated that displacement or HtrA2 proteolytic activity results in the accumulation of unfolded polypeptides within mitochondria, damage or mitochondrial respiration and elevated ROS yield, leading to impairment of mitochondrial bioenergetics [10]. Additionally, depletion of the HtrA2 proteosome leads to transcriptional upregulalion or important nuclear stress responses implicated in mitochondrial PQC mechanisms, including the transcription factor CHOP (CEBP homologous protein), which is differentially regulated upon depletion of this protease [11].
CHOP/DDIT3 comprises 5150 bases of DNA located in the reverse strand region of chromosome 12 and composed of four exons. The gene encodes a 29 kDa protein of 169 amino acid (aa) residues. The protease comprises two functional domains: an N-terminal domain sequence correlated with the transcriptional activation of the protein and a C-terminal basic leucinezipper (bZIP) domain formed by a DNAbinding region of basic amino acids followed by a leucine zip per dimerization domain [12].
Bioinformatics research shows that the bZIP domains of the CHOP protease are highly conserved in humans and in different types of eukaryotic organisms. According to NCBI BLAST comparison, the mouse CHOP and human CHOP (Homo sapiens) protein sequences (obtained from the Protein Data Bank) have 88% identity and 91% similarity along their full length. A Cluslal Omega multiple sequence alignment performed with CHOP protein sequences from different species showed high sequence conservation in the bZIP domains of the analyzed species.
The bZIP domains present in CHOP are structurally similar in their DNA binding and dimerization motifs.
Mitochondria coordinate ATP synthesis through the action of three interlinked processes: oxidative phosphorylation, tricarboxylic acid cycle (TCA) and fatty acid-oxidation (FAO) metabolism. Dysfunction of mitochondrial biogenesis and an increase in oxidative stress result in ATP depletion, which activates glycolysis. Augmentation of this glycolytic pathway can also be a consequence of environmental and genetic mutations [13].
Previous studies have indicated that loss of HtrA2 in mice decreased mitochondrial mass and potential, resulting in ATP depletion [14]. The reduction in ATP synthesis might lead to UPR dysfunction, which is associated with an increase in oxidative stress and is likely to result in further impairment in mitochondria, leading to pathological mechanisms involved in neurodegeneration and apoptosis [15].
In conclusion, the current research supports the role of HtrA2 and CHOP in the transmission of stress signaling and the consequent activation of mitochondrial quality control in Parkinson's disease. Depletion of CHOP function impairs transcriptional activation of mitochondrial quality control signaling. Loss of HtrA2 results in significant up regulation of the molecular quality control marker Hsp60 and organellar quality control marker Atg5. Drug treatment with 6-OHDA induced decreased cell viability and ATP product ions in the HtrA2 KO/CHOP genotype, displaying a drug concentration dependent effect that reflects both the cell viability and the metabolic status of the cells. Drug treatment using 6-OHDA in conjunction with ADEP4 or ACP5 significantly improved cell survival and ATP production, indicating that these drugs might have a protective effect while recovering the effects of 6-OHDA. The findings provide new evidence that depletion of HtrA2 might contribute to an increase in mitochondrial stress and transcriptional up regulation of the nuclear stress response CHOP gene, both of which are involved in Parkinson's disease.
None.
Author declares that there are no conflicts of interest in this work.
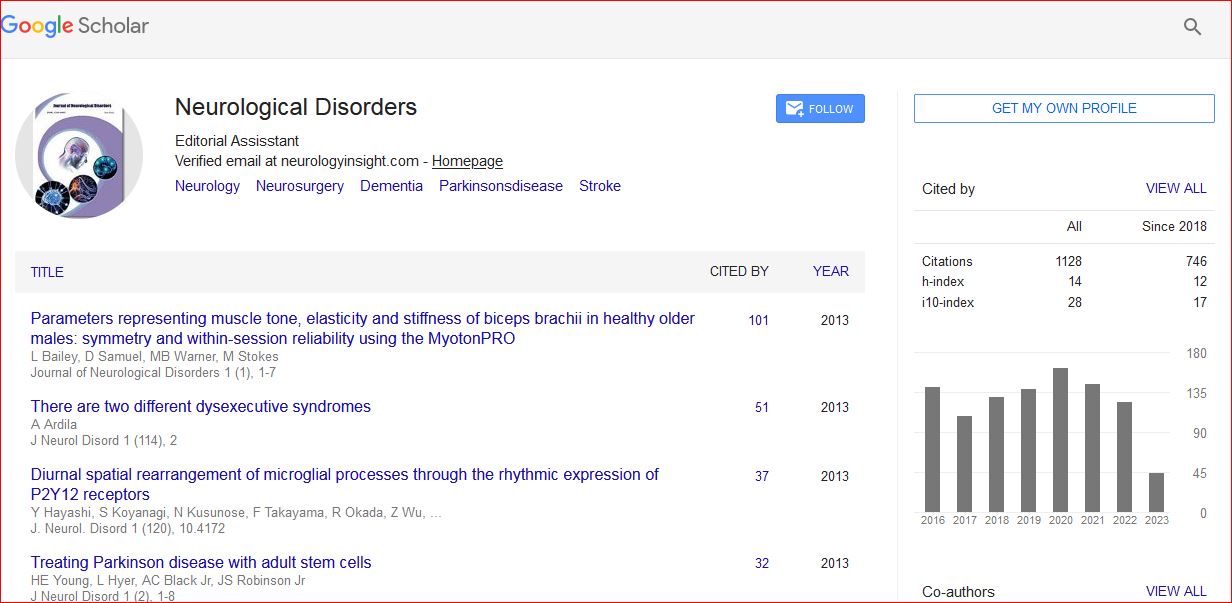
Neurological Disorders received 1343 citations as per Google Scholar report