Review Article - (2023) Volume 9, Issue 1
Received: 17-Apr-2023, Manuscript No. ANTIMICRO-23-96188;
Editor assigned: 20-Apr-2023, Pre QC No. ANTIMICRO-23-96188 (PQ);
Reviewed: 05-May-2023, QC No. ANTIMICRO-23-96188;
Revised: 06-Jul-2023, Manuscript No. ANTIMICRO-23-96188 (R);
Published:
14-Jul-2023
, DOI: 10.37421/2472-1212.2023.9.320
Citation: Sarode, Aditya Prakash and Shraddha Dingare.
"Solid Phase Peptide Synthesis and Its Applications in Tackling
Antimicrobial Resistance." J Antimicrob Agents 9 (2023): 320.
Copyright: © 2023 Sarode AP, et al. This is an open-access article distributed under the terms of the creative commons attribution license which permits unrestricted
use, distribution and reproduction in any medium, provided the original author and source are credited.
Solid Phase Peptide Synthesis (SPPS) is a powerful tool for the design and synthesis of peptides with potential antimicrobial activity. In recent years, SPPS has emerged as a promising strategy for the development of new antimicrobial agents. SPPS is a synthetic method that allows for the efficient and rapid production of peptides using solid-phase supports. This technique involves stepwise addition of protected amino acids to a resin-bound peptide chain, followed by deprotection and cleavage to release the desired peptide. The resulting peptides can be modified to enhance their activity, stability, and bioavailability. One of the key advantages of SPPS is its ability to produce peptides with high purity and homogeneity. This is critical for the development of antimicrobial peptides, which require high levels of activity and specificity to target bacterial cells effectively. Additionally, SPPS allows for the production of peptide libraries, which can be screened to identify new antimicrobial agents with improved activity and selectivity. Several studies have demonstrated the effectiveness of SPPS-derived peptides against multidrug-resistant bacteria, including Methicillin-Resistant Staphylococcus aureus (MRSA), Vancomycin-Resistant Enterococcus faecalis (VRE), and Carbapenem-Resistant Klebsiella pneumoniae (CRKP). These peptides have been shown to target bacterial membranes, disrupt cell wall synthesis, and inhibit essential enzymatic processes. In conclusion, SPPS has emerged as a powerful tool for the development of new antimicrobial agents. The ability to rapidly synthesize and modify peptides with high purity and homogeneity has opened up new opportunities for the design of effective therapies against multidrug-resistant bacteria. As the threat of antimicrobial resistance continues to grow, SPPS will play an increasingly important role in the fight against infectious diseases.
Solid phase peptide synthesis • Lipopeptides • Antibiotics • Methicillin-Resistant Staphylococcus aureus (MRSA) • Purity
Antimicrobial resistance is an increasingly urgent threat to public health worldwide, with the emergence of multidrug-resistant pathogens posing a serious challenge to effective treatment and control of infectious diseases. The development of new antimicrobial agents with novel mechanisms of action is crucial to combat this global health crisis, and solid Phase Peptide Synthesis (SPPS) has emerged as a powerful tool for the design and synthesis of such agents. SPPS is a highly efficient and versatile method for the synthesis of peptides, allowing for the rapid assembly of peptide sequences using solid-phase supports. This technique offers several advantages over other methods of peptide synthesis, including improved purity and homogeneity, reduced side reactions, and greater flexibility in peptide design and modification [1,2]. Moreover, the use of SPPS enables the creation of large peptide libraries for screening against specific targets, facilitating the discovery of novel antimicrobial agents. The potential of SPPS-derived peptides as antimicrobial agents has been extensively studied in recent years. For example, a peptide derived from human lactoferrin was synthesized using SPSS and shown to have potent antibacterial activity against a range of clinically relevant pathogens, including Methicillin-Resistant Staphylococcus aureus (MRSA) and multidrug-resistant Pseudomonas aeruginosa [3]. Similarly, SPPS-derived peptides based on natural antimicrobial peptides such as cathelicidin have been shown to exhibit broad-spectrum activity against a range of bacterial and fungal pathogens [4]. The ability to engineer SPPS-derived peptides for enhanced antimicrobial activity has also been demonstrated in numerous studies. For instance, a designed and synthesized a series of antimicrobial peptides by introducing non-natural amino acids and optimizing peptide sequence, resulting in peptides with improved antibacterial activity against MRSA and Vancomycin-Resistant Enterococcus faecalis (VRE) [5]. Additionally, the use of SPPS has facilitated the production of modified peptides with enhanced stability and bioavailability, which may improve their efficacy as antimicrobial agents. The application of SPPS in the development of antimicrobial peptides has shown promise in preclinical studies, but there are also significant challenges associated with translating these agents to clinical settings. These challenges include issues such as peptide stability and pharmacokinetics, as well as concerns about the potential for bacterial resistance to emerge. Nevertheless, the potential of SPPS-derived peptides as novel antimicrobial agents makes them a promising avenue of research in the fight against antimicrobial resistance. SPPS has emerged as a powerful tool for the design and synthesis of antimicrobial peptides, offering several advantages over conventional methods of peptide synthesis. The ability to engineer peptides for enhanced activity and stability, as well as the potential to create large peptide libraries for screening, makes SPPS a promising approach in the fight against antimicrobial resistance. Further research is needed to address the challenges associated with the translation of these agents to clinical settings, but the potential benefits of SPPS derived peptides in combating multidrug-resistant pathogens make them a promising avenue of research for the future.
Antimicrobial resistance
Antimicrobial Resistance (AMR) is an emerging threat that poses a significant challenge to global public health. It is defined as the ability of microorganisms, such as bacteria, viruses, fungi, and parasites, to resist the effects of antimicrobial drugs, which are commonly used to treat infections. The rapid rise of AMR is a major public health concern, as it has the potential to cause widespread illness and death, particularly in vulnerable populations, such as the elderly, immunocompromised individuals, and children [6].
The rise of AMR can be attributed to a number of factors, including the overuse and misuse of antimicrobial drugs in both human and animal populations, poor infection control practices and the spread of resistant strains of microorganisms through global travel and trade. The misuse of antibiotics is particularly concerning, as it not only leads to the development of resistance but also contributes to the spread of resistant infections. According to a report by the World Health Organization (WHO), AMR is one of the top 10 global public health threats facing humanity. It is estimated that by 2050, the number of deaths attributable to AMR could reach 10 million per year, surpassing the number of deaths caused by cancer. This would have a significant impact on healthcare systems worldwide, as well as the global economy [7].
Mechanism of antimicrobial resistance
• Mutation: Microorganisms can acquire mutations that allow them
to resist the effects of antimicrobial drugs. These mutations can
occur naturally or through exposure to antimicrobial drugs [8].
• Transfer of resistance genes: Microorganisms can acquire
resistance genes from other microorganisms through horizontal
gene transfer, which occurs when genetic material is passed from
one microorganism to another.
• Antibiotic misuse: The overuse and misuse of antibiotics can
lead to the selection of resistant strains of microorganisms.
• Poor infection control: Poor infection control practices, such as
inadequate hand hygiene and improper sterilization of medical
equipment, can contribute to the spread of resistant infections.
Methods to tackle antimicrobial resistance
Tackling AMR requires a multifaceted approach that involves reducing the use of antimicrobial drugs, improving infection prevention and control measures, and developing new antimicrobial drugs [9,10].
• Reducing the use of antimicrobial drugs: Reducing the use of
antimicrobial drugs, particularly antibiotics, is a key strategy in
the fight against AMR. This can be achieved through
improved prescribing practices, such as prescribing antibiotics
only when they are needed, and not using them to treat viral
infections.
• Improving infection prevention and control measures: Improving infection prevention and control measures, such as
hand hygiene, sterilization of medical equipment, and isolation
of patients with resistant infections, can help to prevent the
spread of resistant infections.
• Developing new antimicrobial drugs: Developing new
antimicrobial drugs, particularly those with novel mechanisms of
action, is essential to address the growing threat of AMR. This
can be achieved through increased investment in research and
development, as well as the development of incentives to
encourage the development of new drugs.
• Enhancing surveillance and monitoring: Enhancing
surveillance and monitoring of AMR is essential to track the
emergence and spread of resistant strains of microorganisms.
This can be achieved through the establishment of national and
international surveillance systems, as well as the use of
molecular techniques to identify and track resistant strains.
• Promoting public awareness: Promoting public awareness of
AMR is essential to encourage responsible use of antimicrobial
drugs, improve infection prevention and control measures, and
support the development of new antimicrobial drugs. This can be
achieved through public education campaigns.
Lipopeptide antibiotics
Antibiotics are compounds that are used to treat bacterial infections. They work by killing or inhibiting the growth of bacteria [11]. Lipopeptide antibiotics are a class of antibiotics that are characterized by the presence of a lipophilic tail attached to a peptide moiety. These antibiotics have been used for the treatment of a wide range of bacterial infections, including infections caused by Gram-positive bacteria such as Staphylococcus aureus, Streptococcus pneumoniae, and Enterococcus faecalis. In this review, we will discuss the use of Solid-Phase Peptide Synthesis (SPPS) in the synthesis of lipopeptide antibiotics [12-14].
Categories and classes lipopeptide antibiotics can be divided into two categories: Cyclic and linear. The cyclic lipopeptide antibiotics include daptomycin and its analogs, while the linear lipopeptide antibiotics include polymyxins, colistin, and its analogs.
Cyclic lipopeptide antibiotics: Daptomycin is a cyclic lipopeptide antibiotic that is used to treat infections caused by gram-positive bacteria, including Methicillin-Resistant Staphylococcus aureus (MRSA). Daptomycin works by disrupting the bacterial membrane, leading to cell death. The lipophilic tail of daptomycin allows it to bind to the bacterial membrane, while the peptide moiety inserts into the membrane, causing depolarization and ultimately, cell death. The synthesis of daptomycin and its analogs has been achieved using SPPS. One example of this is the synthesis of the daptomycin analog, triazolyl-daptomycin, which was accomplished using SPPS and click chemistry. In this approach, the peptide was synthesized using Fmoc SPPS, and a triazole linker was introduced using click chemistry [15]. The lipophilic tail was then attached using standard peptide coupling chemistry.
Linear lipopeptide antibiotics: Polymyxins and colistin are examples of linear lipopeptide antibiotics. These antibiotics are used to treat infections caused by gram-negative bacteria, including Pseudomonas aeruginosa and Acinetobacter baumannii. Polymyxins and colistin work by disrupting the bacterial membrane, leading to cell death [16]. The lipophilic tail of these antibiotics allows them to bind to the bacterial membrane, while the peptide moiety inserts into the membrane, causing depolarization and ultimately, cell death. The synthesis of polymyxins and colistin has been accomplished using SPPS. One example of this is the synthesis of colistin using SPPS and fragment condensation. In this approach, the peptide was synthesized using Fmoc SPPS, and the lipophilic tail was synthesized separately. The two fragments were then coupled using fragment condensation.
Solid phase peptide synthesis: A variety of synthetic peptides are important commercial or medicinal goods, ranging from the sugar replacement dipeptide aspartame to therapeutically utilised hormones such as oxytocin, adrenocorticotropic hormone, and calcitonin. This module gives an introduction to the topic of synthetic peptides and proteins. It goes through how to choose a solid support and typical coupling reagents. Additional information on frequent side reactions and the synthesis of changed residues is provided [17].
Following the introduction of solid-phase methods, peptide synthesis became a more realistic aspect of modern scientific study. The idea behind Solid-Phase Peptide Synthesis (SPPS) is to keep the chemistry that has worked in solution but add a covalent attachment step that connects the nascent peptide chain to an insoluble polymeric substrate (resin). Following that, the attached peptide is expanded through a series of further cycles. The core of the solid-phase technique is that reactions are accelerated by the use of excess soluble chemicals, which can be eliminated by simple filtration and washing with no manipulative losses. The crude peptide is released from the support after chain elongation is complete. Solid-phase synthesis is a typical method for producing peptides. In the SPPS technique, peptides are typically synthesized from the carbonyl group side (C-terminus) to the amino group side (N-terminus) of the amino acid chain, despite the fact that peptides are physiologically synthesized in the opposite manner in cells. An amino-protected amino acid is bonded to a solid phase material or resin (most typically, low cross-linked polystyrene beads) in peptide synthesis, establishing a covalent bond between the carbonyl group and the resin, most commonly an amido or ester linkage [18]. The amino group is then de-protected and interacts with the carbonyl group of the next N-protected amino acid. A dipeptide is now present in the solid phase.
The amino group is then de-protected and interacts with the carbonyl group of the next N-protected amino acid. A dipeptide is now present in the solid phase. The solid phase now contains a dipeptide. This process is repeated until the desired peptide chain is created. Once all reactions have been performed, the peptide is cleaved off the bead.
9-Fluorenylmethyloxycarbonyl (Fmoc) and t-Butyloxycarbonyl (Boc) are the most often used protective groups for amino groups in peptide synthesis. Several amino acids have functional groups on their side chains that must be prevented from interacting with the N-protected amino acids entering the system. They must be stable during peptide synthesis, unlike Boc and Fmoc groups, albeit they are also removed during peptide deprotection [19]. This approach removes unreacted compounds by washing without causing product loss. The peptide chain is formed from the carboxyl end of the peptide's amino terminus. The carboxyl moiety of each incoming amino acid is activated by one of many processes and interacts with the amino group of the preceding amino acid. The amino group of the entering residue is momentarily blocked to prevent peptide bond formation at this location. The residue is unblocked at the start of the next synthesis cycle. Furthermore, protecting groups are added to the reactive side chains of amino acids. The peptide chain grows longer when the synthesis cycle is repeated. Excess reagents are used to accelerate reactions as much as possible. As a result, the finished product has the maximum potential yield.
The side-chain protecting groups are removed when the peptide has been completely formed, and the peptide is cleaved off the solid support under conditions that do no injury to labile residues. The outcome is then evaluated to validate the sequence. The peptide is typically purified using gel chromatography or HPLC. The blocking group utilised to block the amino group determines both the type of side-chain protecting groups and the synthesis chemistry used. The two most often used amino protecting groups are Fmoc (9-Fluorenyl-methoxycarbonyl) and tBoc (tert.-Butyloxycarbonyl) [20]. Tert.-butanol ester, ether, and urethane derivatives protect the Fmoc side chain, whereas benzyl alcohol ester, ether, and urethane derivatives protect the tBoc side chain. The latter are typically modified for increased acid stability by the addition of electron-withdrawing halogens. Also employed are cyclopentyl or cyclohexyl alcohol ethers and ester derivatives. Fmoc, the protective group, is base-labile. To remove it, a dilute base, such as piperidine, is frequently used. The treatment with Trifluoroacetic Acid (TFA) eliminates the side-chain protecting groups while also cleaving the bond that holds the peptide to the support. To dissolve the tBoc protecting group, a mild acid (usually dilute TFA) is utilized. To deprotect the amino acid side chains and dissolve the peptide from the resin support, Hydrofluoric Acid (HF) can be utilized. Fmoc is a milder approach than tBoc since the peptide chain is not exposed to acid at each cycle [21].
Chemicals and materials
Resins: Solid-phase synthesis, which includes SPPS and SPOS, requires a solid support that is persistently bonded to a linker, allowing for permanent anchoring of a target peptide to it. One of the most frequent resins used in SPPS is 1-2% divinylbenzene-cross-linked polystyrene. Other examples include the following: Polyethylene glycol-polystyrene graft solid supports, such as PEG-PS, Champion support, and Tenta gel; polyamide solid supports; beaded Polyethylene Glycol-Acrylamide (PEGA) copolymers; and Polyethylene-Polystyrene (PE-PS) films. The reader is invited to refer to the following review for alternate polymeric solid supports (Table 1).
| Resin | Structure |
|---|---|
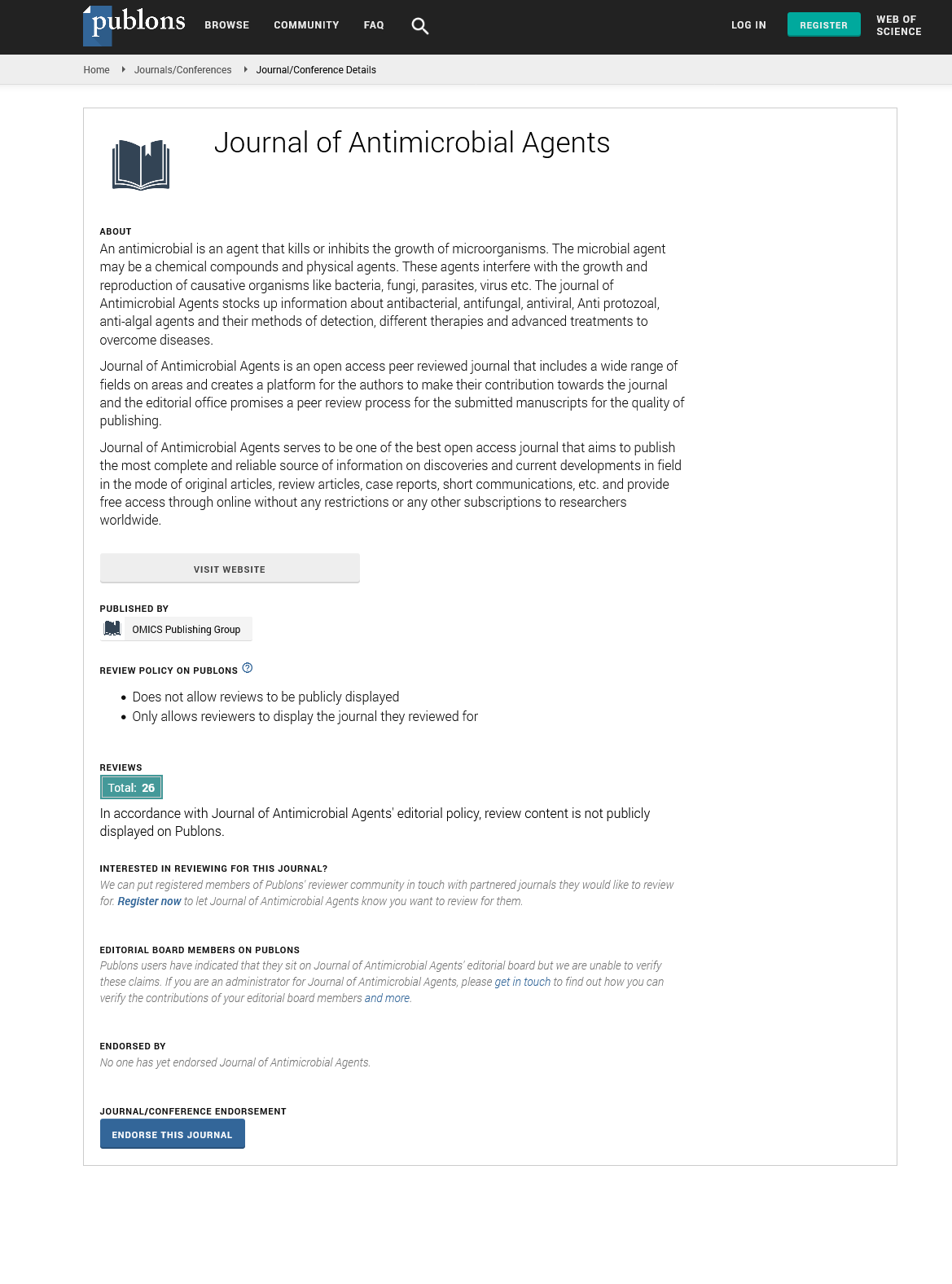
| 2-Chlorotrityl chloride resin | 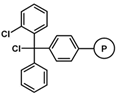 |
| 4-Methyl Benzhydryl amine (MBHA) | 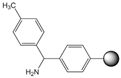 |
| Polystyrene | 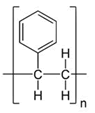 |
Table 1. Commonly used resins and their structure.
Coupling agent: The EDCHCl coupling reaction should be performed under basic conditions in the presence of additives such as N-Hydroxy-5-Norbornene-2,3-Dicarboximide (HONB), ethyl-2-cyano-2-(hydroxyimino) acetate (OxymaPure), 3-sulfo-N-hydroxysuccinimide (sulfo-HOSu), and others. Furthermore, the following stand-alone coupling reagents were used: 2-(5-norbornene-2,3-dicarboximido)-1,1,3,3-Tetramethyluronium Tetrafluoroborate (TNTU); and (1-cyano-2-ethoxy-2-oxoethylidenaminooxy) dimethylamino-morpholino-carbenium hexafluorophosphate (COMU) (Table 2).
| S. no. | Coupling agent | Structure |
|---|---|---|
| 1. | HATU | 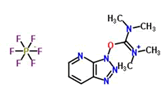 |
| 2. | HBTU | 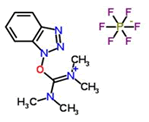 |
| 3. | HCTU | 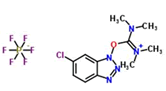 |
| 4. | TNTU | 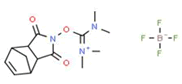 |
| 5. | DMT | 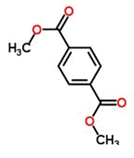 |
Table 2. Commonly used coupling agents and their structure.
Stability of peptides
Peptide stability is determined by the amino acid sequence and content. In general, lyophilized peptides are more stable than those in solution. Peptides stored in lyophilized form at -20°C or -80°C are less likely to degrade. If possible, avoid exposure to pH more than 8 and to ambient oxygen. Peptide degradation routes include the following:
Hydrolysis: It is a common issue for peptides containing Asp (D). Avoid sequences that contain the amino acid combinations Asp-Pro (D-P) or Asp-Gly (D-G), since they may dehydrate, resulting in a cyclic imide intermediate. Sequences containing Ser (S) may produce cyclic imide intermediates that can break the peptide chain to a lesser extent.
Oxidation: The most common amino acids that undergo reversible oxidation are Cys (C) and Met (M). Cysteine oxidation is accelerated at higher pH levels because the thiol is more easily deprotonated and forms intra or inter-chain disulfide bonds more quickly. Treatment with dithiothreitol (DTT) or Tris (2-Carboxyethyl Phosphine) Hydrochloride (TCEP) can reverse disulfide bonds. Methionine residues oxidise by both chemical and photochemical routes, resulting in methionine sulfoxide and/or methionine sulfone, which are difficult to reverse.
Diketopiperazine and pyroglutamic acid formation: Formation of diketopiperazine and pyroglutamic acid: Diketopiperazine is formed when Glycine (G) is in the third position from the N-terminus, and even more so when Pro (P) or Gly (G) is in position 1 or 2. A nucleophilic assault of the N-terminal nitrogen on the amide carbonyl between the second and third amino acids results in the cleavage of the first two amino acids as a diketopiperazine. If Gln (Q) is at the N-terminus of the sequence, pyrogalactic acid production is nearly unavoidable. To generate a deaminated pyroglutamyl peptide analogue, the N-terminal nitrogen attacks the side chain carbonyl carbon of the Gln (Q). Conversion may also occur in peptide sequences with Asn (N) at the N-terminus.
Mechanism
Boc/Bzl protection: Boc protecting groups are utilized to temporarily shield the N alpha nitrogen groups of amino acids in the Boc/Bzl protection scheme, whereas benzyl-based protecting groups give more permanent side-chain protection. Because both the Boc and benzyl-based protecting groups are acid labels, Boc/Bzl is not a genuine orthogonal protection system. It is practical, however, since the Boc group may be removed under mild circumstances (50% TFA in DCM), whereas benzyl-based protective groups require extremely strong acids, such as HF or TFMSA, to be removed. AAPPTec provides Boc-protected amino acids at reasonable prices for peptide synthesis.
Boc deprotection mechanism: Tert-butyl carbonium ions are produced during Boc-deprotection, as demonstrated in the process below. These cations can then react with nucleophiles to generate isoprene or tert-butyl adducts. Within a peptide, tryptophan (Trp), Cysteine (Cys), or Methionine (Met) residues can combine with tert-butyl carbonium ions to form unwanted peptide side products (Figure 1).

Figure 1. BOC protection strategy.
By adding 0.5% Dithioethane (DTE) to the cleavage solution, the tert-butyl cations are scavenged and the generation of peptide side products is prevented. The deprotected amine is in the form of a TFA salt after the Boc group has been removed by TFA treatment. Before the next amino acid can be linked, the salt must be converted to free amine. Typically, the resin-peptide is treated with a 50% solution of Diisopropylethylamine (DIEA) in Dichloromethane (DCM), followed by multiple washes. Castro and colleagues reported employing a BOP/DIEA in situ neutralization technique. Kent and Alewood pioneered in situ neutralization using HATU or HBTU coupling [22]. In situ neutralization can increase coupling yields when aggregation presents issues, in addition to saving time by removing separate neutralization and washing operations. Because aggregation occurs mostly in the neutral resin-peptide, in situ neutralization should reduce aggregation by reducing the amount of time the deprotected resin-peptide is in the neutral state.
Fmoc/tBu protection: The alpha nitrogen of the amino acids is protected with the base labile Fmoc group in this scheme, while the side chains are protected with acid labile groups based on the tert-butyl protecting group or the trityl (triphenylmethyl) group. Because the side chain protective groups may be removed without displacing the N-terminal protection, this is an orthogonal protection scheme. It is useful when side chains must be specifically changed, for as when the peptide is labeled or cyclized through the side chain. AAPPTec provides a wide range of Fmoc-protected amino acids.
FMOC deprotection mechanism: When a base abstracts the comparatively acidic proton from the fluorenyl ring system, the Fmoc group is eliminated, resulting in Î2-elimination and the creation of dibenzofulvene and carbon dioxide. Dibenzofulvine is a reactive electrophile that would rapidly and irreversibly connect to the deprotected amine if not scavenged. Secondary amines, such as piperidine, are added to dibenzofulvene to reduce negative side effects. As a result, piperidine is commonly employed to remove the Fmoc group while simultaneously scavenging the dibenzofulvene by-product [23]. Describes the use of a 5% piperidine solution to remove Fmoc protective groups from resin bound amino acids. The time necessary to remove the Fmoc group from the first five alanine residues in the creation of a poly-alanine peptide was between 20 and 30 minutes. The deprotection period increased to 100 to 170 minutes for the following five alanine residues (Ala 6 through Ala 10), most likely due to aggregation. Piperidine deprotection of three minutes or less is used in recently published optimized rapid Fmoc methods [24]. The absorbance of the dibenzofulvene adduct is monitored by certain automated peptide synthesizers, such as the Fmoc XC series, and the time for the deprotection process is automatically extended until no additional adduct production is observed.
The Fmoc protective group is removed significantly faster by 1,8-Diazabicyclo-1-2 undec-7-ene (DBU) than by piperidine [25]. When Fmoc deprotection is sluggish or partial during peptide synthesis, substituting piperidine with DBU can enhance deprotection yield and hence increase peptide yield [26]. Piperidine is frequently added to react with the fulvulene byproduct since DBU is non-nucleophilic and will not react with it. DBU should not be employed when Aspartic Acid (Asp) residues are present in the peptide-resin because DBU catalyzes aspartimide production followed by piperidine reaction (Figure 2).

Figure 2. General mechanism of synthesis of peptide via solid phase peptide synthesis.
Applications
SPPS (Solid-Phase Peptide Synthesis) is a strong and frequently used method for producing peptides and peptide-based medicines. To make a peptide chain, amino acids are progressively assembled onto a solid substrate, generally a resin. SPPS has been widely employed in the synthesis of a wide range of medications, including antibacterial, anticancer, antiviral, and metabolic problems. In this essay, we shall describe the uses of SPPS in drug production, as classified by these criteria [26].
Antimicrobial drugs: Antimicrobial pharmaceuticals are medications that are used to treat infections caused by bacteria, fungus, viruses, or parasites. Peptide-based antibiotics have garnered popularity as prospective antibiotic replacements due to their ability to overcome antibiotic resistance. Several lipopeptide antibiotics, including daptomycin, a lipopeptide antibiotic used to treat gram-positive bacterial infections, have been synthesised utilising SPPS. SPPS has also been utilised to create colistin, a cyclic polypeptide antibiotic used to treat multidrug-resistant gram-negative bacterial infections (Figures 3 and 4) [27].

Figure 3. Daptomycin.

Figure 4. Colistin.
Anticancer drugs: Because of their selectivity and minimal toxicity, peptides have showed promise as anticancer medicines. SPPS has been utilised to synthesise anticancer peptides such as somatostatin analogues, which have been employed in the treatment of neuroendocrine tumours. Enfuvirtide, a peptide-based antiviral medication that has also been demonstrated to have anticancer potential [28], is another example.
Antiviral drugs: Because of the discovery of viral fusion peptides that impede viral entrance into host cells, peptide-based antiviral medicines have been produced. Enfuvirtide, a peptide-based antiviral medication, has been used to treat HIV. SPPS has been utilized to make enfuvirtide and other peptide-based antiviral medicines, such as brincidofovir, a peptide pro-drug used to treat cytomegalovirus infections (Figure 5).

Figure 5. Brincidofovir.
Metabolic disorders: SPPS has also been employed in the synthesis of peptide-based medicines, which are used to treat metabolic diseases including diabetes and obesity. SPPS has been used to synthesise Glucagon-Like Peptide-1 (GLP-1) analogues for the treatment of type 2 diabetes. A GLP-1 analogue, liraglutide, has been authorised for the treatment of type 2 diabetes and obesity. Exenatide, a GLP-1 analogue used to treat type 2 diabetes, is another example. Overall, SPPS has shown to be a powerful technique for the synthesis of a wide range of peptide-based medications. Because of its flexibility, efficiency, and convenience of use, it has become a preferred approach for producing peptides and peptide-based therapeutics (Figure 6) [29].

Figure 6. Liraglutide.
In conclusion, Solid-Phase Peptide Synthesis (SPPS) has revolutionized the field of peptide-based drug synthesis. SPPS offers numerous advantages, such as high yield, purity, and ease of use, making it a popular choice for the synthesis of peptides and peptide-based drugs. This review article discussed the applications of SPPS in the synthesis of various drugs belonging to different categories, including antimicrobial, anticancer, antiviral, and metabolic disorders. Antimicrobial resistance is a growing public health concern worldwide, with the emergence of multidrug-resistant strains of bacteria. Peptide-based antibiotics synthesized using SPPS have shown promise in overcoming antibiotic resistance, particularly lipopeptide antibiotics such as daptomycin and colistin. However, continued research is required to address the challenge of increasing antimicrobial resistance. In summary, SPPS has proven to be a valuable tool for the synthesis of various peptide-based drugs in different categories. As drug resistance continues to pose a threat to public health, peptide-based drugs synthesized using SPPS offer a promising alternative to conventional antibiotics and other drugs. Further research and development in the field of SPPS and peptide-based drugs are necessary to address the challenge of increasing antimicrobial resistance and to improve patient outcomes.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Journal of Antimicrobial Agents received 444 citations as per Google Scholar report