Perspective - (2022) Volume 11, Issue 3
Received: 03-Mar-2022, Manuscript No. jmmd-22-64798;
Editor assigned: 07-Mar-2022, Pre QC No. P-64798;
Reviewed: 14-Mar-2022, QC No. Q-64798;
Revised: 17-Mar-2022, Manuscript No. R-64798;
Published:
23-Mar-2022
, DOI: 10.37421/2161-0703.2022.11.341
Citation: Alther, Sherman. “Systems Microbiology to Functional Genomics.” J Med Microb Diagn 11 (2022): 341.
Copyright: © 2022 Alther S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
The field of functional genomics attempts to describe the functions and interactions of genes and proteins by making use of genome-wide approaches, in contrast to the gene-by-gene approach of classical molecular biology techniques. It combines data derived from the various processes related to DNA sequence, gene expression, and protein function, such as coding and noncoding transcription, protein translation, and protein DNA, protein RNA, and protein–protein interactions. Together, these data are used to model interactive and dynamic networks that regulate gene expression, cell differentiation, and cell cycle progression. Studying cells at a systems level has been facilitated by recent technological advancements, as well as the availability of complete genome sequences. Since the landmark publication of the first draft of the human genome in the genomes of hundreds of organisms from all branches of the tree of life have been sequenced [1,2].
This has led to improved annotations of genes and their products and has enabled genome-wide studies aimed at understanding interactions and molecular processes in the cell. DNA microarrays consist of thousands of microscopic DNA spots probes that are bound to a solid surface, such as glass or a silicon chip Affymetrix or microscopic beads Illumina. Labelled single-stranded DNA or antisense RNA fragments from a sample of interest are hybridized to the DNA microarray under high-stringency conditions. Each probe is identified by its location on the DNA microarray and the amount of hybridization detected for a specific probe is proportional to the level of nucleic acids from the corresponding genomic location in the original sample Over the years, sequencing pipelines have greatly improved in throughput and costs for instruments and reagents, along with improvements in computational power, data storage and bioinformatics tools that facilitate the analysis of the growing quantities of sequence reads [3].
Together, these advancements have caused a dramatic drop in sequencing costs, Several new companies, such as Helices Biosciences, Pacific Biosciences, Technologies, are currently developing novel, so-called third generation sequencing techniques that do not require amplification of template DNA, but are able to read the sequence of single DNA molecules. These technologies could significantly advance the sequencing field by greatly reducing the cost for reagents and improving the throughput, while simultaneously eliminating any bias introduced during the template amplification step of the NGS protocol mass spectrometer consists of three components: an ion source to convert a gas-phase sample into ions, a mass analyser to separate the ions by means of an electromagnetic field and a detector. The development of ionization techniques that enable the transfer of proteins and peptides into the gas phase without substantial degradation has been crucial for the application of mass spectrometry MS in large-scale proteomic studies. The most commonly used ionization techniques are matrixassisted laser desorption ionization and electrospray ionization [4,5].
These ionization techniques can be combined with various types of mass analysers that separate ions based on the mass-to-charge ratio by either trapping ions in an electrical field trapping mass spectrometers or by accelerating ions through an electrical field and measuring the time-of-flight. A comparison of instrument configurations that are most commonly used in proteomics is provided elsewhere. The most advanced mass spectrometer available to date is the Orbit rap, which has a high resolution, a high mass accuracy, and a large dynamic range that make it suitable for a wide range of proteomics and metabolomics applications. He most common strategy for proteomic studies is a bottom-up approach, in which a protein sample is first enzymatically digested into smaller peptides, followed by separation of the peptides by charge, hydrophobicity, or a combination of these characteristics, and then injected into the mass spectrometer.
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
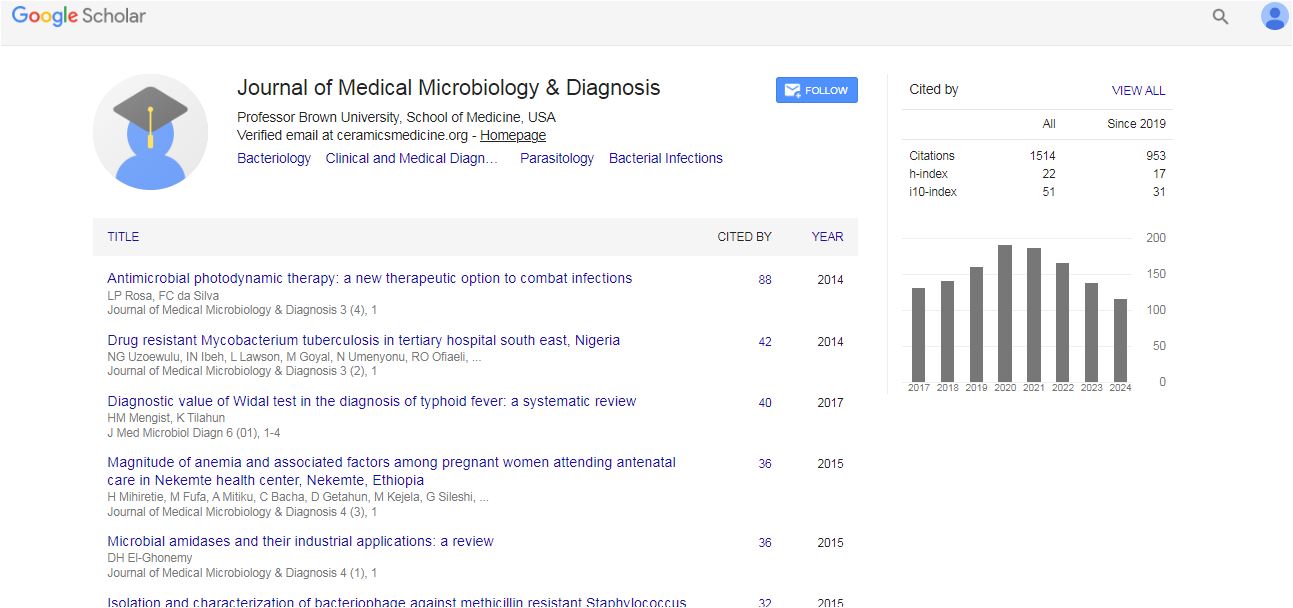
Medical Microbiology & Diagnosis received 14 citations as per Google Scholar report