Perspective - (2022) Volume 10, Issue 4
Received: 01-Apr-2022, Manuscript No. jaat-22-65047;
Editor assigned: 03-Apr-2022, Pre QC No. P- 65047;
Reviewed: 15-Apr-2022, QC No. Q- 65047;
Revised: 19-Apr-2022, Manuscript No. R- 65047;
Published:
26-Apr-2022
, DOI: 10.37421/ 2329-6542.22.10.206
Citation: Baczek, Tomasz. “Two Well-known Displaying Strategies
(AM1 and DFT) were utilized to Foster Models.” J Astrophys Aerospace Technol
10 (2022): 206
Copyright: © 2022 Baczek T. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
While developing Quantitative Structure-Retention Relationships (QSRR) models it is crucial to construct a data matrix, consisting of both experimentally designated values (the ones that are being modelled) and molecular descriptors, representing physicochemical properties in form of numbers. In order to designate those descriptors, studied compounds need to be modelled and optimized using molecular modelling methods.
From a variety of possible choices, semi-empirical AM1 (being an often discredited method due to the fact that it is quick and provides various approximations) and DFT (being a complex, quantum mechanics-based method, which is more time-consuming yet still bases on approximations) are one of the most popular [1]. In this study one took a set of carefully chosen compounds used to model retention on C18 column, and modelled their molecules using both of those techniques, which were afterwards subjected to Genetic-Algorithm Multiple Linear Regression (GA-MLR) in order to derive and compare achieved models and their parameters. Study found that for the tested chromatographic system (C18 column and a set of model fifteen compounds) there is no advantage in using DFT over AM1, despite common modelling principles [2].
What is more important, the developed models are accurate and use very simple descriptors, which can be easily calculated without any need for complex 3D modelling of structures. The derived models enabled also to assess the physicochemical properties of the tested column finding slight similarities and dissimilarities between the applied models [3]. Quantitative Structure-Properties Relationship (QSPR) studies are a popular way of understanding not only how chemical structure impacts retention but also in developing models that help predict certain values as well as properties of the chromatographic systems different than retention times. Additionally, approaches such as Quantitative Retention-Activity Relationships (QSAR) were developed, where retention in a certain stationary phase directly mimics physiological conditions, allowing predicting drug activity [4]. In order to create such a model, apart from experimentally designated values a set of molecular descriptors, calculated from geometrically optimized compound model and representing physicochemical properties in a numerical way, must be provided. Only a matrix containing both of those data allows to study correlation between value of interest (e.g., retention time) and independent values (molecular descriptors), unravelling before-mentioned relationships between structure (and properties of chemicals) and their retention (expressed for example as retention time) [4].
The quality of this matrix and how well it represents real-life molecules is strictly linked with models used to calculate descriptors. Two-dimensional flat models of chemicals will generally provide less data than three-dimensional models, with their energy optimized according to quantum chemistry principles. As such, two very popular methods to optimize molecules have emerged -AM1, based on semi-empirical approach, which is fast to perform on personal computer (yet provides some approximations that often led to it being discredited), and Density Functional Theory (DFT) type, based on quantum mechanics, which is time and resource consuming, but can provide better approximations and representations of certain chemical properties [5]. The aim of this study was to compare AM1 and DFT methods on a set of 15 classic modelling compounds elaborated during the studies on chromatographic column characterization and retention predictions with the use of QSRR and use of genetic-algorithm multiple linear regressions (GA-MLR) to construct QSRR models.
Over the years, GA-MLR has emerged as better technique than classical MLR, allowing to create models that not only have good parameters, but also are interpretable and suitable for possible further optimization of the development of HPLC separations, involving the applications of pharmaceutical and biomedical analysis. What is more, in recent years a few studies showed potential hidden in both AM1 and DFT modelling techniques, proving that in order to gain a full spectrum of information a one method should rarely be used. Additionally, DFT method was shown to not always provide better calculations over semi-empirical methods, while AM1 was even shown to be one of the less optimal methods to use in certain cases. Using GA-MLR might probably allow finding new conclusions regarding older studies, and help to find whether DFT in certain datasets provides any real advantage over AM1.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
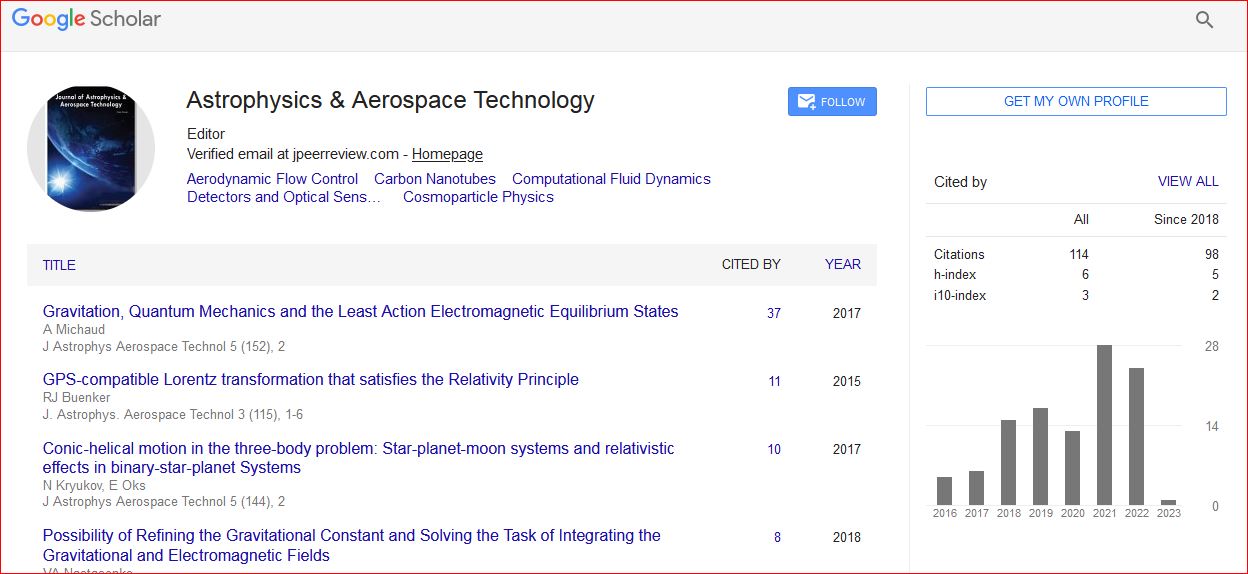
Astrophysics & Aerospace Technology received 114 citations as per Google Scholar report