Opinion - (2023) Volume 14, Issue 1
Received: 02-Jan-2023, Manuscript No. jpm-23-90422;
Editor assigned: 04-Jan-2023, Pre QC No. P-90422;
Reviewed: 16-Jan-2023, QC No. Q-90422;
Revised: 21-Jan-2023, Manuscript No. R-90422;
Published:
28-Jan-2023
, DOI: 10.37421/2090-0902.2023.14.411
Citation: Dubiel, Javier. “Using Atomic Coordinates as the Basis, Machine Learning Classifiers for Magnetism.” J Phys Math 14 (2023): 411.
Copyright: © 2023 Dubiel J. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
One of the most prominent quantum phenomena, magnetism of materials, is used in a wide range of functional applications, including data storage, highresolution imaging, spintronic devices, and magnetism of materials. High-energy scientific instruments Particular types of magnetism are thought to be associated with unusual quantum phases like topological superconductivity and high-Tc. Large materials' spatial correlations between magnetic moments offer a wide variety of possible magnetic configurations, in contrast to small molecules, whose magnetic structures only contain a few high- and low-spin configurations. With an infinite number of wavevector, moment, and correlation length combinations, quantum spin liquids spin glass can create structures such as antiferromagnetism, non-collinear magnetism, and skyrmions. Therefore, it is essential to determine the magnetic structures, either experimentally or theoretically, for the purposes of material discovery and technological advancement in general [1].
The most recent experimental method, neutron scattering, as well as resonant X-ray scattering, have made it possible to identify magnetic structures at the atomic level. However, these measurements, which necessitate largescale neutron sources or synchrotron X-ray radiation, are severely constrained by the available beam time and capacity. According to the most comprehensive database, only 1,500 materials' magnetic structures have been identified through these experimental spectra since the 1950s. Therefore, unless the capacity of these facilities is increased by orders of magnitude, a pure experimental exploration of magnetic materials cannot yet meet the rapidly increasing demand for the discovery of new magnetic materials [2].
Theoretically, the magnetism of small molecules has been accurately predicted through ab initio simulations made with cutting-edge methods from quantum chemistry and physics. However, it is impractical to apply it to large materials beyond the nanoscale without any approximation due to the exponential expansion of the Fock space with system size. The first-principles DFT simulations and the corrections that go along with them provide an efficient means of achieving a balance between precision and scalability. Due to the absence of static correlation and the delocalization error, the magnetic moment and correlations may be underestimated. DFT-based methods have made high-throughput calculations on more than 10,000 materials possible, allowing for preliminary statistical predictions of the properties of materials. Even when compared to experiments and wave function-based approaches, the computational complexity of DFT calculations remains negligible, preventing the discovery of chemical compositions in a vast, possibly infinite parameter space [3].
Due to the fact that electronic structure theory evaluates the energy for a specific electronic configuration, which includes the magnetic structure, the standard simulation also requires traversing all configurations for a single atomic structure and determining the ground-state magnetic configuration. A "guessing-computing" duo, or guessing a large number of configurations before computing each one individually, is the result of the large number of possible magnetic configurations. As a result, the majority of computational effort is spent on irrelevant magnetic excited states rather than working on the actual ground states. If the ground-state magnetic structure could be accurately predicted, highthroughput calculations would be significantly sped up, moving us one step closer to simulation-free material discovery [4].
Due to the difficulties in determining its structure through calculations and experiments, the use of machine learning to improve magnetic structure determination has recently received a lot of attention. In some recent studies, DFT calculations and machine learning have been combined some of which incorporate machine learning models into the "guessing" phase of the guessingcalculating procedure. For instance, machine learning has been utilized to narrow the search space for potential magnetic configurations in the "guessing" step. With this strategy, the primary calculation task continues to be performed using the standard first-principles DFT calculations. Some other works employ model Hamiltonians primarily classical spin models, and machine learning methods to fit the model's free parameters, such as from experimental data containing spin. All things considered, the immediate prediction of attractive design from simpler nuclear construction, also referred to as replacing the "processing" step, is still uncertain.
A thorough explanation of magnetism can be challenging, according to Rodriguez-Carvajal and Villain. We focus on two distinct descriptions with few variables in this work: labels of magnetic order and propagation vectors Magnetic ordering labels like ferromagnetic (FM) and antiferromagnetic (AFM) are useful because they break down the complexity of magnetic structures into classes that are relevant to particular applications and easy to understand. Both ferrimagnetic and ferromagnetic materials exhibit a spontaneous magnetization: when there is no external magnetic field present, a net magnetic moment that is not zero; however, while all of the magnetic dipoles in FM point in the same direction, some of them point in the opposite direction in FiM. In antiferromagnetic materials, dipoles that point in opposite directions in a regular pattern cancel each other out, resulting in zero net magnetic moment. Because the orientation of the magnetic dipoles in non-magnetic materials is irregular and void of pattern, the net magnetic moment is zero. A propagation vector is a vector in reciprocal space that describes the presence of magnetic order and symmetry breaking (ibid.). A non-zero propagation vector is one indication of a more intricate magnetic structure, which goes beyond the FM, AFM, and NM ternary classifications. Even though these descriptions are expressive, they are not all-encompassing; subsequent work will provide descriptions of magnetic order that are more indepth [5].
The amount of times each element in the training set appears. It is simple to identify correlations between large numbers of training samples containing certain elements, such as Mn, Fe, Co, Ni, and Cu, and high accuracies of those elements. From the point of view of data abundances, this helps us gain a deeper comprehension of the various accuracies that exist across various elements. On the other hand, the elements with lower prediction accuracies, such as Ga, Lu, and Re, tend to be less common. However, it is essential to note that some rare earth elements, such as Tb, Dy, and Ho, perform exceptionally well despite the small number of training samples. This is because rare earth elements frequently exist alongside other abundant elements; For instance, Mn, Fe, Mo, Co, and Ni are found in 65.9% of Tb, Dy, and Ho structures.
None.
None.
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
Google Scholar, Crossref, Indexed at
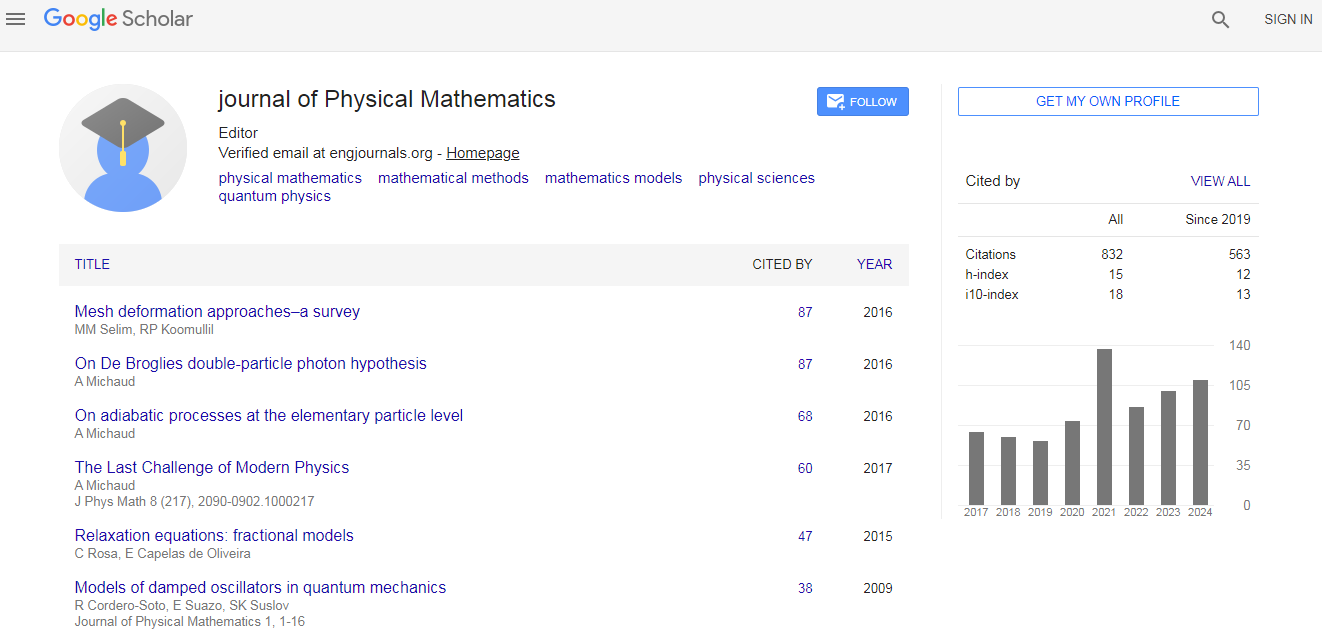
Physical Mathematics received 686 citations as per Google Scholar report