Dr Omid Ahmad Faizi
French Medical Institute for Children, Afghanistan
Scientific Tracks Abstracts: Journal of Clinical Case Reports
Gaucher’s disease is one of the most common lysosomal storage diseases characterized by hematologic abnormalities, organomegaly, and skeletal involvement. It is caused by reduced activity of the enzyme acid β-glucosidase which is encoded by a gene on chromosome 1q21-q311. The enzymatic defect results in the accumulation of glycolipid substrates, mainly glucosylceramide, in cells of the macrophage-monocyte system. It is one of the most prevalent genetic defects among Ashkenazi Jews2. There are 3 clinical subtypes distinguished by the presence or absence and progression of neurologic manifestations: type 1 or the adult, non neuronopathic form; type 2, the infantile or acute neuronopathic form; and type 3, the juvenile or subacute neuronopathic form3. Here we report a case of Gaucher’s disease that presented with pancytopenia and splenomegaly. We present this case to emphasize the importance of considering storage disorders like Gaucher’s disease when evaluating a case of unexplained pancytopenia and organomegaly even in adults. A 20 year old female presented with generalised weakness and easy fatigability, menorrhagia and dragging sensation in the left upper quadrant of abdomen. There was no history of fever, rashes, joint or bone pain, yellowish discoloration of eyes and urine, night sweats or weight loss. There was no history of similar illness in her family members which included her parents and one sibling and there was no consanguinity among parents. On physical examination, the patient had pallor but no icterus or lymphadenopathy. She had massive splenomegaly with firm consistency, regular margin and smooth surface and a non tender, mild hepatomegaly. There was no sign of any neurological deficit and the rest of the systemic examination was normal. Lab investigations showed pancytopenia (hemoglobin=6.8 g/dl, white blood cells=2.45x109/L and platelets=40x109/L). Examination of peripheral smear showed severe anemia with marked anisopoikilocytosis with microcytic hypochromic blood picture with moderate leukopenia and marked thrombocytopenia. Corrected reticulocyte count was 1.6%. Liver and kidney function tests were normal. Prothrombin time was 14s [international normalized ratio(INR)=1.14]. Serum Iron was 30mcg/dl with total iron binding capacity = 474mcg/dl suggesting chronic blood loss. Ultrasonography confirmed massive splenomegaly (23cm) and mild hepatomegaly (15cm) with normal portal vein diameter and no evidence of splenic vein thrombosis. Montoux test was negative and Erythrocyte sedimentation rate was normal. Serological markers for HIV, Hepatitis B and Hepatitis C were negative. Antinuclear antibody, malaria antigen test and rK 39 antibody were also negative. To evaluate the cause of pancytopenia, bone marrow biopsy was done which revealed hypercellular marrow with clusters of ovoid macrophages with abundant fibrillary cytoplasm and round eccentric nucleus (gaucher cells). To confirm the diagnosis of Gaucher’s disease (Type 1), β-glucosidase enzyme assay by fluorometry method was done which showed β-glucosidase level of 0.51 nmol/hour/ml (normal value >2 nmol/hour/ml). Final diagnosis was G.D (type-1).
Dr Omid Ahmad Faizi is working as a researcher at French Medical Institute for Children in Afghanistan.
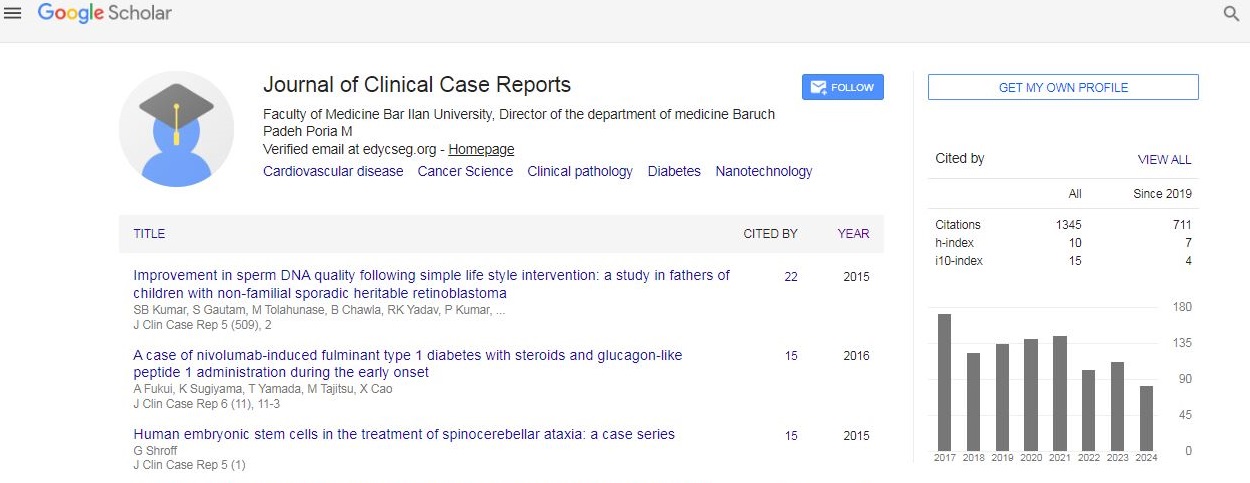
Journal of Clinical Case Reports received 1345 citations as per Google Scholar report


 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 