Asif Mahmood
Pfizer, USA
Posters & Accepted Abstracts: J Bioanal Biomed
A fundamental regulatory requirement of biosimilarity is that there are no clinically meaningful differences between the biologic product (i.e. biosimilar) and the reference product in terms of quality, safety, and efficacy. Patient safety is a key consideration throughout the step-wise development of a biosimilar that continues through post-marketing safety monitoring. Because biologic products, including monoclonal antibodies, by their very nature are capable of eliciting immune responses in humans; immunogenicity is a focus of safety assessments during development. An immune response may lead to altered efficacy or compromised safety. Specific effects of immune responses include an immune response that may include immune complex formation resulting in decreased or increased clearance of the biologic and neutralization of the activity of the biologic. Changes in manufacturing, purification, or packaging, as well as shipping and storage conditions, have also the potential to impact the molecular structure of the biologic product and, hence, its immunogenic potential. Additionally immunogenicity also can be impacted by the dose, formulation, and route of administration, as well as individual patient factors such as atopy and immunosuppression. Manufacturing & Safety: Because even small alterations in the source materials or production process of any biologic product may lead to changes in molecular structure, and potentially its biologic effects, manufacturing processes should be carefully controlled at each step. During development of the biosimilar, it is critical that not only the primary amino acid sequences but also higher-order structures be reproduced to the greatest extent possible. This is an important consideration for the biosimilar manufacturer given the proprietary (and usually confidential) nature of the original manufacturing process of the reference product, as well as subsequent changes. Nevertheless, even manufacturers of reference products may encounter significant batch-to-batch variations. Safety also can be compromised by process-related impurities from cell substrates (e.g., host cell DNA and host cell proteins), cell culture components (e.g., antibiotics and media components), and downstream processing steps (e.g., reagents, residual solvents, endotoxins, and bioburden). Formulation, Packaging & Safety: Formulation differences of the biosimilar relative to the reference product are permitted provided the manufacturer of the biosimilar submits evidence demonstrating the differences are not clinically meaningful. Clinically meaningful differences could include a difference in the expected range of safety, purity, and potency between the biosimilar and reference product, whereas slight differences in rates of occurrence of adverse events would not be considered clinically meaningful, and even could occur between studies of the same reference product.
E-mail: Asif.Mahmood@pfizer.com
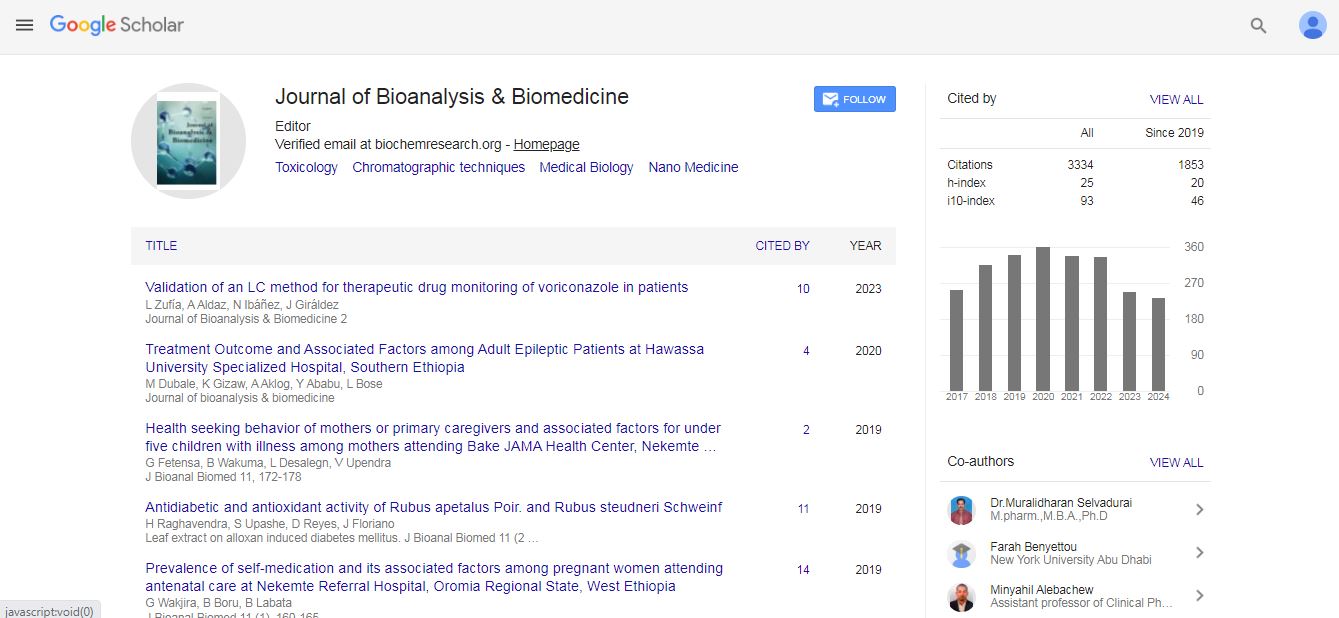
Journal of Bioanalysis & Biomedicine received 3099 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 