Ali Radhi and Kamran Behdinan
University of Toronto, Canada
Scientific Tracks Abstracts: J Material Sci Eng
External loads and stimuli are known to change crystalline structures with distinct structural symmetry groups. With this change, the crystalline structure should be identified through its atomic bonding represented in a large number of observed deformation mechanisms along with its embedded defects for various crystal systems. Information extracted should discern ideal crystal phases from other generated crystalline features during atomistic simulations for multiple crystalline groups. Ideally, a method for arbitrary structures is preferred for ease of transferability between multiple crystalline systems case studies. The common neighborhood parameter (CNP) method has an optimal approach towards structural characterization through combining the advantages of both the common neighbor analysis (CAN) and the centro-symmetry parameter (CSP) methods. A new method will be presented as an improved approach on structural analysis from predominance of the common neighbor over the current atom, termed predominant common neighborhood parameter (PCNP). A more innovative method will also be presented that is based on the cumulative distance of nearest neighbors. This method rewards atoms with perfect surrounding structures with higher parametric values instead of smaller ones, labeled as cumulative common neighborhood parameter (CCNP). These methods are ideal to characterize cross-atomic species interactions in a more elaborate crystalline systems and centrosymmetric space groups. The methods showed higher sensitivity than CNP for characterizing atomic features during deformation. Enhancements to the methods can be included by considering larger range of atomic interactions present in the second neighborhood locale.
Ali Radhi has expertise in multi-scale modeling of ceramic polymorphs and structural characterization of deformation mechanics for non-monoatomic systems. His work is based on atomistic to continuum bridging approaches for high levels of coupling during atomic simulation for optimal use of molecular dynamics approaches with fraction of the computational costs. He has built a foundation for such modeling through extensive work with algorithmic optimization with mathematics department and going through clinical attachments in hospitals in Kuwait.
Email: ali.radhi@mail.utoronto.ca
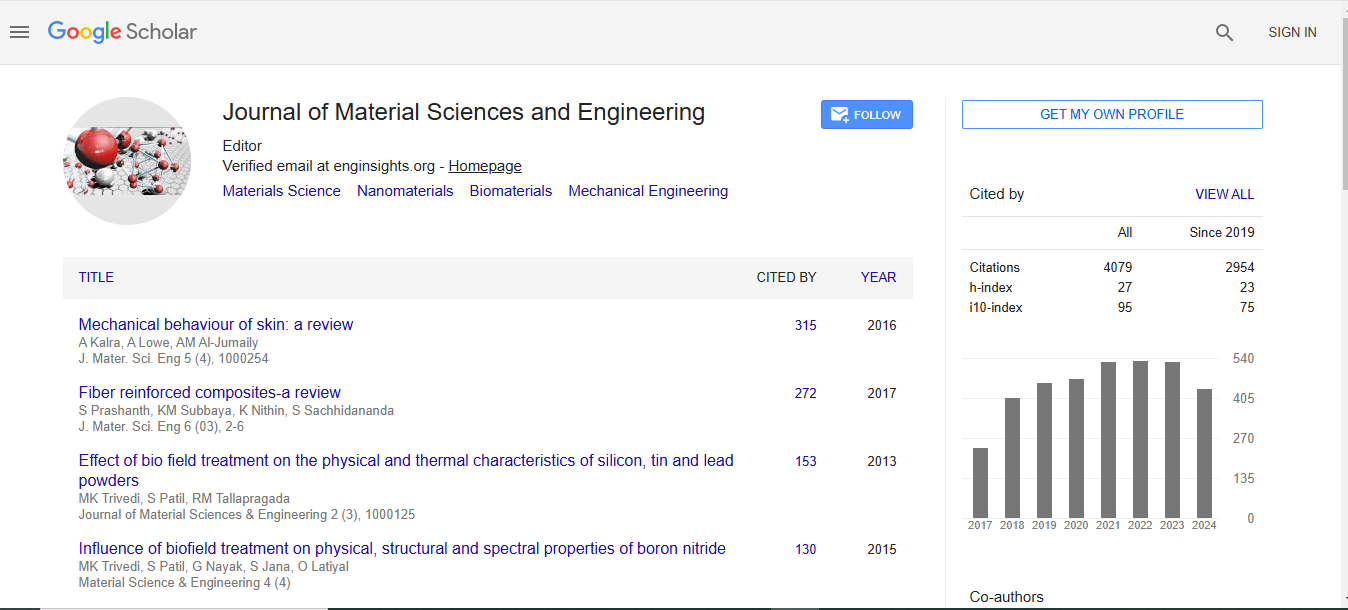
Journal of Material Sciences & Engineering received 3677 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 