Eric Dybeck
Pfizer Global Research and Development, USA
Keynote: Pharmacoeconomics
Many pharmaceutical compounds can crystallize into more than one solid form with varying chemical and physical properties. Selecting a solid form that is stable, manufacturable, and bioavailable is therefore a critical step in the drug development process, and an unintended late-stage transformation into a new solid form can severely delay a therapeutic reaching the market. Computational models of a compoundâ�?�?s solid form landscape represent an inexpensive method to reveal previously unobserved crystal structures and assist in selecting the appropriate form to advance to commercial manufacturing. However, conventional approaches for crystal structure prediction (CSP) often neglect the effects of temperature and entropy on solid form stability, and the consequences of this assumption are not fully understood. Molecular Dynamics simulations were used here to introduce the effects of temperature and entropy into models of small molecule crystal structures. These simulations were used to estimate how solid form stability changes with temperature and more generally elucidate the consequences of temperature and entropy effects on the predicted polymorphic landscape. These simulations demonstrate that large entropy differences can exist between enantiotropically related polymorphs, and molecular dynamics is capable of capturing these temperature effects on thermodynamic stability in agreement with experiments. In addition, the dynamic simulations reveal when a crystal undergoes an order-disorder transition that can facilitate the stabilization of a higher-energy form over a lower-energy form at nonzero temperature. Finally, the results provide evidence for a long standing hypothesis that multiple lattice minima can exist within the same ambient-temperature free energy basin. The inclusion of temperature and entropy effects in future CSP models has the potential to significantly boost the accuracy and efficiency of predicting the crystals of pharmaceutical compounds. Recent Publications 1. Dybeck E, Abraham N, Schieber N and Shirts M (2017) Capturing entropic contributions to temperature-mediated polymorphic transformations through molecular modeling. Crystal Growth and Design. 17(4):1775-1787. 2. Dybeck E, Schieber N, Shirts M (2016) Effects of a more accurate polarizable Hamiltonian on polymorph free energies computed efficiently by reweighting point-charge potentials. JCTC. 12(8):3491-3505. 3. Dybeck E, K�?¶nig G, Brooks B and Shirts M (2016) Comparison of methods to reweight from classical molecular simulations to QM/MM Potentials. JCTC. 12(4):1466-1480. 4. Dybeck E, Plaisance C and Neurock M (2017) Generalized temporal acceleration scheme for kinetic Monte Carlo simulations of surface catalytic processes by scaling the rates of fast reactions. JCTC. 13(4):1525-1538. 5. Hibbitts D, Dybeck E, Lawlor T, Neurock M and Iglesia E (2016) Preferential activation of CO near hydrocarbon chains during Fischer-Tropsch synthesis on Ru. Journal of Catalysis 33:91-101.
Eric Dybeck has a background in Chemical Engineering and Computational Chemistry in both academic and industrial environments. His research interests include crystal structure prediction, computational materials science, and heterogeneous catalysis. In his graduate work, he developed a novel method to compute the effects of temperature on the stability of organic small molecule crystal structures. He now works for Pfizer where he applies similar computational methods on active pharmaceutical development projects.
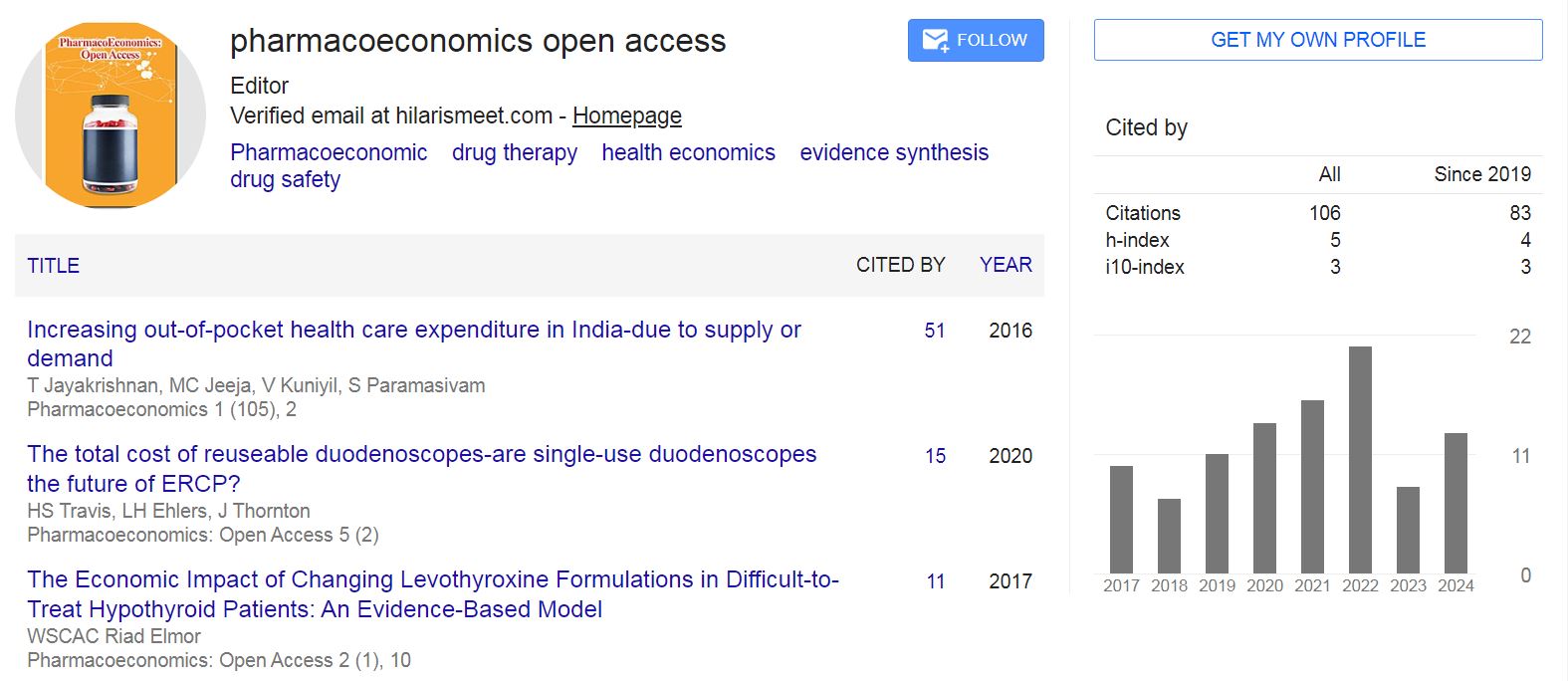
Pharmacoeconomics: Open Access received 106 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 