Andres Gonzalez-Hernandez and Yhojan Diaz
Universidad Industrial de Santander, Colombia
Posters & Accepted Abstracts: J Material Sci Eng
Group IV transition metals Ti and Zr are the most attractive metallic materials in aerospace and nuclear industries. They have high specific strength, good biocompatibility, excellent corrosion and oxidation resistance and low neutron-capture. Ti and Zr have three polymorphic modifications, including high pressure Ï�?-phase with hexagonal structure. In this work, the high-pressure behavior in the crystal structure of ZrTi2 have been systematically studied by using universal structure prediction method together with density functional theory. The structure prediction was carried out using USPEX (Universal Structure Predictor: Evolutionary Xtallography) code with the Vienna Ab-initio Simulation Package (VASP) code for 0, 25, 50, 75, and 100 GPa. Uspex was developed by Oganov, Glass, Lyakhov and Zhu, which allows one to predict the most stable crystal structure and a number of low-energy metastable structures for a chemical composition at any pressure conditions without requiring any experimental input. In this work, the pseudopotentials of Ti and Zr elements were treated their respective orbitals 3p64s23d2 and 4s24p65s24d2 as valence electronic configurations. For USPEX, the maximum total numbers of atoms in the unit cell were limited to 6 (2 atoms of Zr and 4 atoms of Ti). According to the previous studies [6], three possible structures were chosen to study with USPEX: �?±-ZrTi2 with hcp structure (P63/mmc, 194), �?±-ZrTi2 with hcp structure (P6/mmm, 191), and �?²-ZrTi2 with bcc structure (Im3m, 229). Additionally, three structured obtained with same software were used to refine the search for stable structures: P6mm (183), P4mcm (127) and P-3m1(164). The results showed that the space groups 183, 164 and 194 are the most stable structures at zero temperature and pressure for ZrTi2. The structure �?±-ZrTi2 (194) is the most stable between 3 and 75 GPa. Finally, the space group 164 is the most stable between 75 and 100 GPa. Recent Publications: 1. Oganov A.R., Glass C.W. (2006). Crystal structure prediction using ab initio evolutionary techniques: principles and applications. J. Chem. Phys. 124, art. 244704. 2. Oganov A.R., Lyakhov A.O., Valle M. (2011). How evolutionary crystal structure prediction works - and why. Acc. Chem. Res. 44, 227-237 3. Lyakhov A.O., Oganov A.R., Stokes H.T., Zhu Q. (2013). New developments in evolutionary structure prediction algorithm USPEX. Comp. Phys. Comm. 184, 1172-1182 4. Zhang W.W., Oganov A.R., Goncharov A.F., et al. (2013). Unexpected stoichiometries of stable sodium chlorides. Science 342, 1502-1505. 5. Bili�?�?, A., Gale, J. D., Gibson, M. A., Wilson, N., & McGregor, K. (2015). Prediction of novel alloy phases of Al with Sc or Ta. Scientific Reports, 5, 9909. 6. Yuan, XL., Xue, MA., Chen, W. et al. (2016). A first-principle study on the phase transition, electronic structure, and mechanical properties of three-phase ZrTi2 alloy under high pressure. Eur. Phys. J. B 89, 246.
Andres Gonzalez-Hernandez has his expertise in material science. Ph.D, full time professor at the Metallurgical and Material Science at the Department at Universidad Industrial de Santander, Bucaramanga, Colombia. He received his B.Sc. in Metallurgical Engineering in the Universidad Pedagógica y Tecnológica de Colombia, Tunja, Colombia in 2004, his M.Sc. Engineering degree in 2008 and his Ph.D in Engineering at Universidad de Antioquia, in 2014 together with the degree Ph.D. in Ceramic Materials in Université de Limoges, France. His research interests include: thermal spraying coatings, thermal barrier coatings, crystal structure prediction, first-principles calculation, DFT, Uspex.
Email:andresggonzalezh@gmail.com
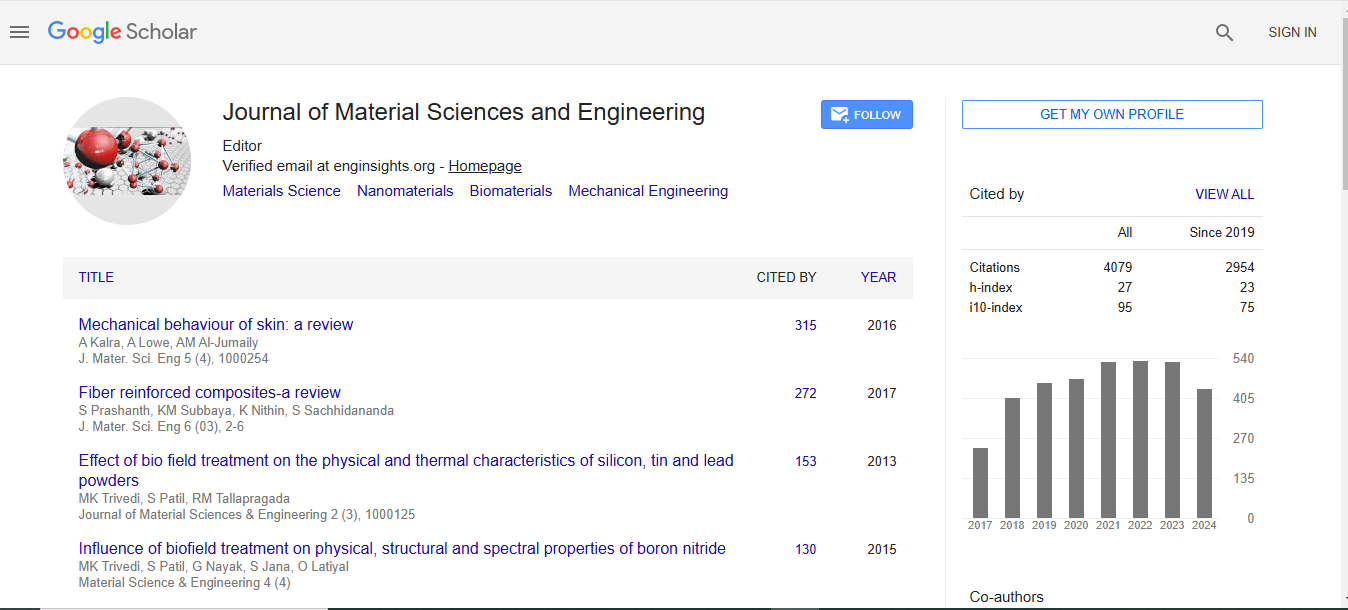
Journal of Material Sciences & Engineering received 3677 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 