Sergei Y Funikov, Alexander P Rezvykh, Maria M Chicheva, Ekaterina A Vikhareva, Mikhail B Evgen��?ev and Aleksey A Ustyugov
Engelhardt Institute of Molecular Biology-RAS, Russia
Institute of Physiologically Active Compounds-RAS, Russia
Scientific Tracks Abstracts: J Neurol Disord
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder leading to the eventual death of motor neurons. Described
cases of familial ALS have emphasized the significance of protein misfolding and aggregation of two functionally related
proteins, FUS and TDP-43, implicated in RNA metabolism. Here in, using the in vivo model of FUS-mediated proteinopathy
(�?�?FUS1-359 mice) we performed the comprehensive analysis encompassing the onset of the first clinical symptoms inclusions
formations as well as changes in gene expression profile in motor neurons and surrounding microglia. The obtained data
enable to conclude that FUS-mediated proteinopathy is virtually asymptomatic in terms of both the clinical symptoms and
molecular aspects of neurodegeneration until it reaches the terminal stage of disease progression (120 days age). From this time
point the pathological process develops very rapidly resulting in massive FUS-positive inclusions formation accompanying the
transcriptional burst in the spinal cord cells. Specifically, it manifests in activation of pro-inflammatory phenotype of microglial
cells and malfunction of acetylcholine synapse transmission in motor neurons. Overall, we assume that a highly reproducible
course of the pathological process, as well as described accompanying features, make �?�?FUS1-359 mice a convenient model for
testing potential therapeutics against proteinopathy-induced decay of motor neurons.
Recent Publications
1. Hardiman O, et al (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17071.
2. Shang Y and E J Huang (2016) Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res.
1647:65-78.
3. Philips T and J D Rothstein (2015) Rodent models of amyotrophic lateral sclerosis. Curr Protoc Pharmacol. 69:1-21.
4. Shelkovnikova TA, et al (2013) Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA
binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J Biol Chem. 288(35):25266-74.
5. Farrawell N E, et al (2015) Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular
inclusions. Sci Rep. 5:13416.
Sergei Y Funikov has completed his PhD at Engelhardt Institute of Molecular Biology-Russian Academy of Sciences. His main research focuses on studying the regulation of small non-coding RNAs (in particular, microRNAs and Piwi-interacting RNAs) in stress response as well as investigation of the immune and neuroprotective properties of human recombinant heat shock protein 70 (Hsp70). His study concerns the molecular aspects underlying the progression of motor neurons failure on the model recapitulating the clinical symptoms of amyotrophic lateral sclerosis.
E-mail: sergeifunikov@mail.ru
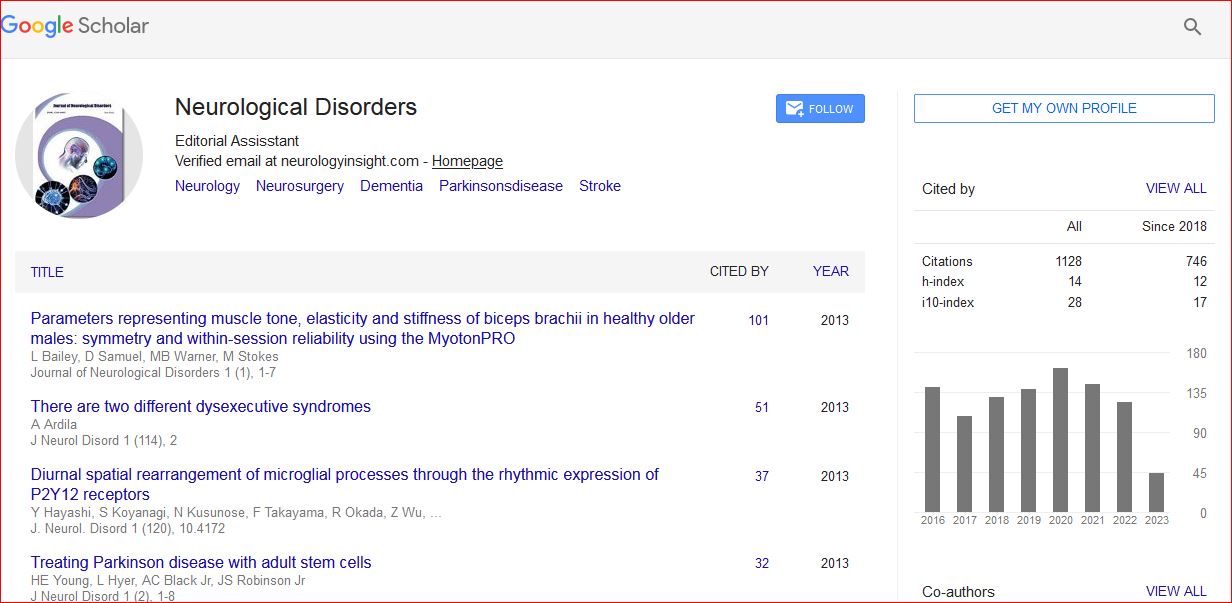
Neurological Disorders received 1343 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 