Karen L Zuleta Hernandez, Marly D Olarte Verdugo, Kevin N Chacon and Sol M Mejia
Universidad Jorge Tadeo Lozano, Colombia
Pontificia Universidad Javeriana, Colombia
Posters & Accepted Abstracts: J Material Sci Eng
This research focused on the computational study of intermolecular interactions responsible for the geometric preferences and stability of (ethanol)8-water heterononamers. The B3LYP functional was used as implemented in Gaussian 09. The potential energy surface was explored using the ab-initio molecular dynamics method (ADMP) and a stochastic method (Simulated Annealing) to find starting structures that were optimized with the B3LYP/6-31+G (d) approximation; obtaining 9 stable heterononamers. After reoptimizing those structures including dispersion correction (D3) and a larger base, B3LYP-D3/6-311++G(d,p), the number of stable structures was reduced to 7. The major structural changes were different orientations of the alkyl chains. It was calculated that the most stable heterononamer (Hnon-I) has an isomeric population of 98%. From this structure were designed and optimized homologous structures with only ethanol and with only methanol molecules, as well as a (methanol)8-water structure. Measurements made including geometric, energetic, and topological data. The binding energy (nonamerization) is the difference between the energy of the heterononamer and the sum of the energy of each isolated monomer. In the same way other state functions were calculated (�?H, �?S, and �?G). As an example, the following are the energetic data of Hnon-I: �?E=-303.68 kcal/mol; �?H=-339.29 kcal/mol; �?S=275.32 kcal/K*mol, and �?G=4.16 kcal/mol. These results imply that the formation of Hnon-I is a highly exothermic and non-spontaneous process. It was found that the cycles formed by O-H---O interactions are fundamental for stabilizing the (hetero) nonamer regardless of their nature. Weaker interactions (C-H---O and H---H) were revealed by means of the molecular graphs calculated through the topological analysis of electron density according to the Quantum Theory of Atoms in Molecules.
Karen L Zuleta H was born in Bogotá in 1997. She is a student at the Jorge Tadeo Lozano University in the Chemical Engineering program. She is currently doing her research in theoretical and computational chemistry that involve molecular structure, energetic, and binding.
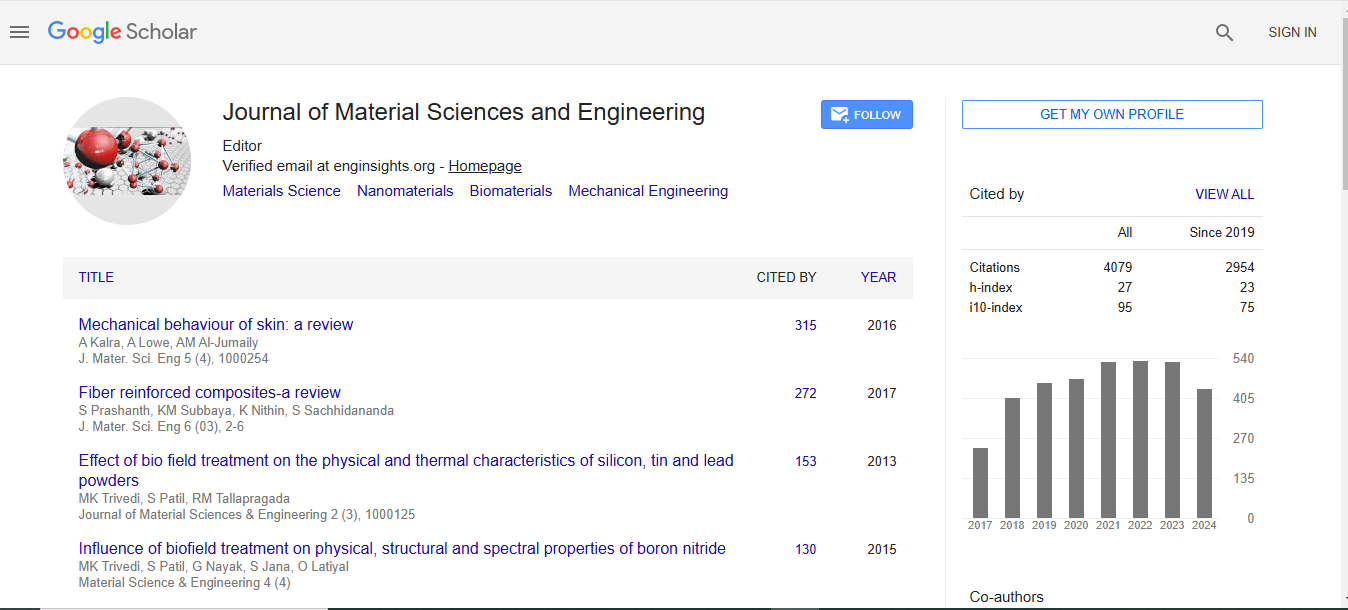
Journal of Material Sciences & Engineering received 3677 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 