Samiksha Gupta
Educe Solutions, India
Posters & Accepted Abstracts: J Formul Sci Bioavailab
The process of informing adverse drug reaction to the regulatory authorities or too concerned persons is known as reporting. It is mandated by drug regulatory authorities for a marketing authorization holder to report adverse event reports. Different countries have their reporting rules. The case reports should include information on adverse event, suspected and concomitant product therapy details, patient details, clinical course and outcomes, narrative, dechallenge and rechallenge information, company comments etc. The events should be coded with the most recent version of MedDRA used at the lowest level term (LLT) and in line with the latest points to consider document. The follow up information received after the submission of initial version of the case should also be reported to the regulatory authorities depending upon medical significance of the information received. Significant follow up would require reporting to the authority whereas non-significant follow up information would not. Care should be taken for the multiple cases reported in a single report e.g., multiple cases available in a single publication, mother baby case report. These reports should be linked. Upon completion of data entry in a case report, the case can be either reported as a paper format (CIOMS-I) or electronically (E2B). Case series should be developed and evaluated by the MAHs for any signal or safety information. Different parameters to considered for case evaluation are Temporal relationship, causality assessment, other confounding factors etc. On the basis of evaluation, a safety issue should be communicated to the regulatory authority as well as general public.
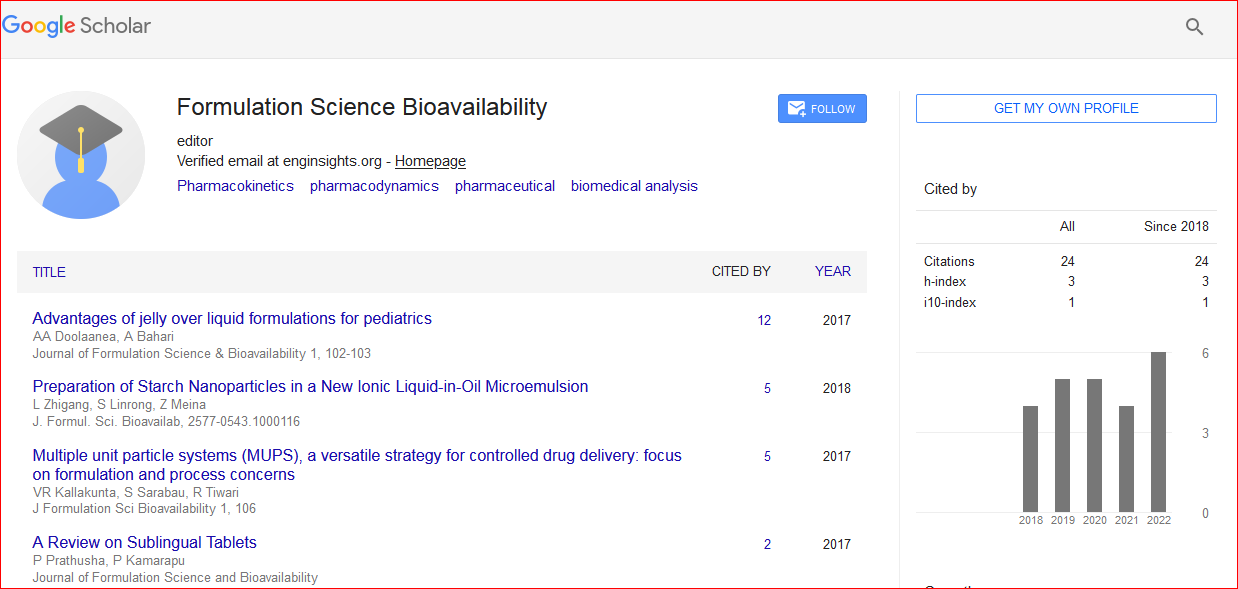
Journal of Formulation Science & Bioavailability received 23 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 