Shuji Ogata and Takahiro Tsuzuki
Nagoya Institute of Technology, Japan
Posters & Accepted Abstracts: J Material Sci Eng
The lithium ion battery (LIB) has become an indispensable energy storage device for various electronic systems such as PC. Expecting wider-spread usage of LIBs, the system of quinone molecules encapsulated in the single-wall carbon nanotube (SWCNT), which is free from rare-metals as Co, has been proposed as a next-generation cathode electrode material. Note that the dissolution of quinone molecules into liquid electrolyte is suppressed by the encapsulation. In this talk we report our large-scale (containing thousands of atoms) first-principles simulation results about the structure of quinone molecules and their dynamics in adsorbing and releasing of Li-ions in SWCNT. Since substantial charge-transfer exists between quinines and SWCNT, we should perform the simulation at the electronic level instead of using a classical inter-atomic potential. Unique features of our simulation code is following the real-space grid implementation of the density-functional theory (RGDFT), in which the finite difference method is used for derivatives of the Kohn-Sham orbitals and Hartee field, is known to have attractive points of parallelizability and applicability to various boundary conditions in addition to universality in target materials. Taking the divide-and-conquer strategy we have recently developed the linear-scaling, divide-and-conquer-type RGDFT (DC-RGDFT) code for large-scale simulation with short computation timings. The empirical VdW potential called DFT-D2 is added to DC-RGDFT. Following the experimental conditions, 9, 10-phenanthrenequinone (PhQ) is considered. We will report about the relaxed PhQ structures in SWCNT with or without Li-ions. We also simulate the atomic dynamics at adsorption and releasing of Li-ions.
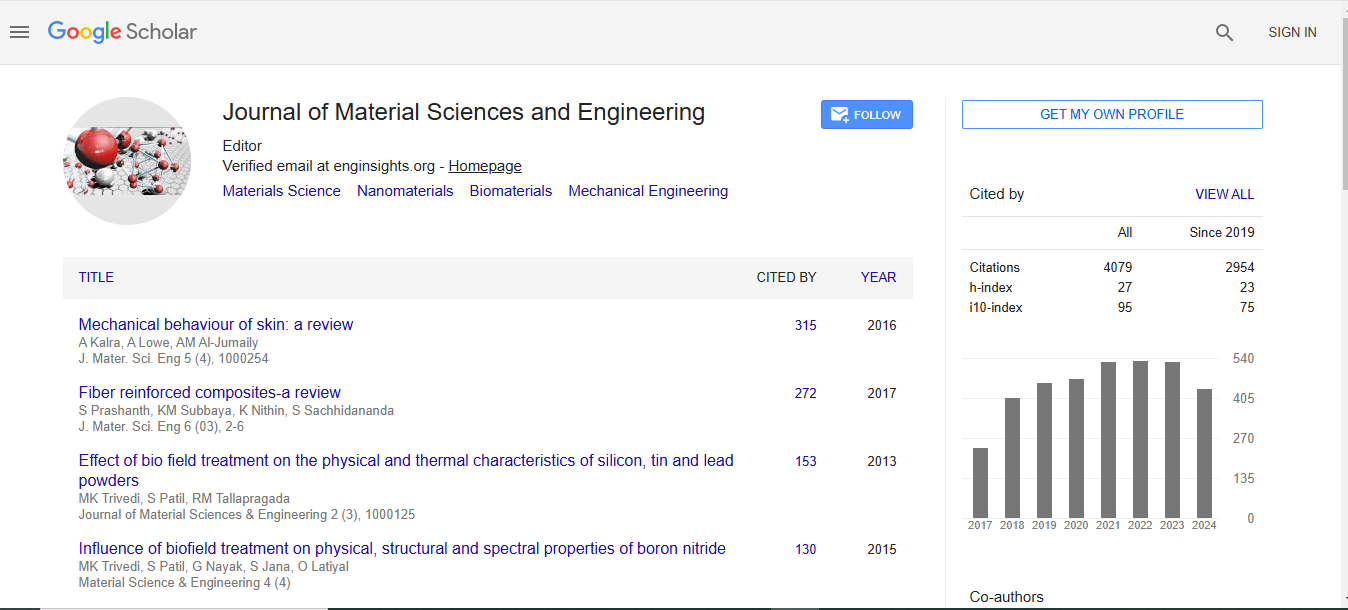
Journal of Material Sciences & Engineering received 3677 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 