Daniel Zielinski
Posters: J Comput Sci Syst Biol
Genome-scale metabolic network reconstructions provide a context within which omics data can be analyzed to understand phenotypic functions. Here, we develop a workflow to integrate three disparate data types (metabolomic, fluxomic, and thermodynamic) within the context of a metabolic reconstruction. First we determine that the data sets are thermodynamically consistent. Then, we use the mass action stoichiometric simulation (MASS) model-building framework to develop three condition-specific kinetic models of E. coli core metabolism. The results from this study demonstrate: 1) that the MASS approach, that generates condition-specific rate constants based on in vivo data, can generate network-scale dynamic models in a data-driven manner, 2) that the MASS models generated under many conditions have underlying dynamic similarities, and 3) that this structured and integrated omics data analysis yields consistent physiological results from data that otherwise would be treated as a series of independent measurements. The impending onslaught of quantitative in vivo metabolomics data can thus be converted into useful dynamic models in a data-driven fashion to generate descriptions of integrated network functions.
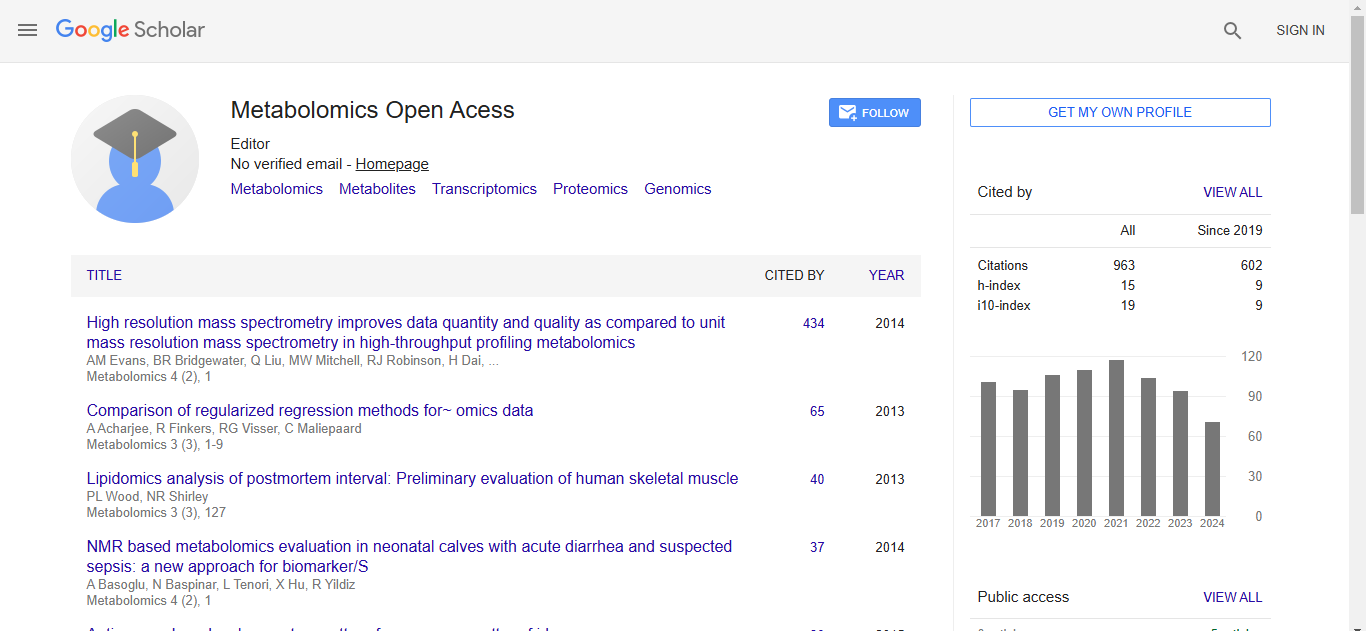
Metabolomics:Open Access received 895 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 