Graham S Jackson
UCL Institute of Neurology, UK
Posters & Accepted Abstracts: J Mol Biomark Diagn
Prion diseases are a group of untreatable and transmissible neurodegenerative conditions of humans and animals caused by the misfolding of prion protein (PrP). Transmissibility of disease between humans and animals is a key defining feature of the group, and has resulted in several high profile epidemics of acquired prion disease, including for example, kuru in humans and bovine spongiform encephalopathy (BSE) and its human counterpart variant Creutzfeldt-Jakob disease (vCJD). Prion diseases also occur more commonly without any obvious exposure to an infectious source, the typical clinical syndrome being a rapidly progressive dementia termed sporadic CJD. The diagnosis of CJD is typically made late in the clinical course by which time patients are in an advanced state of neurological decline. Pathology is detectable in many tissues, particularly those of the central nervous system but the invasive nature and risks associated with diagnostic tonsillar or brain biopsy, preclude investigation in the early stages of illness. The infectious agent itself is largely or solely comprised of a misfolded and aggregated conformer of the normal cellular prion protein which can catalyse the conversion of further PrP to disease associated forms by a templating process. Detection of these abnormal forms of PrP is the main focus of strategies to develop new early diagnostics in human biofluids.
Email: g.jackson@prion.ucl.ac.uk
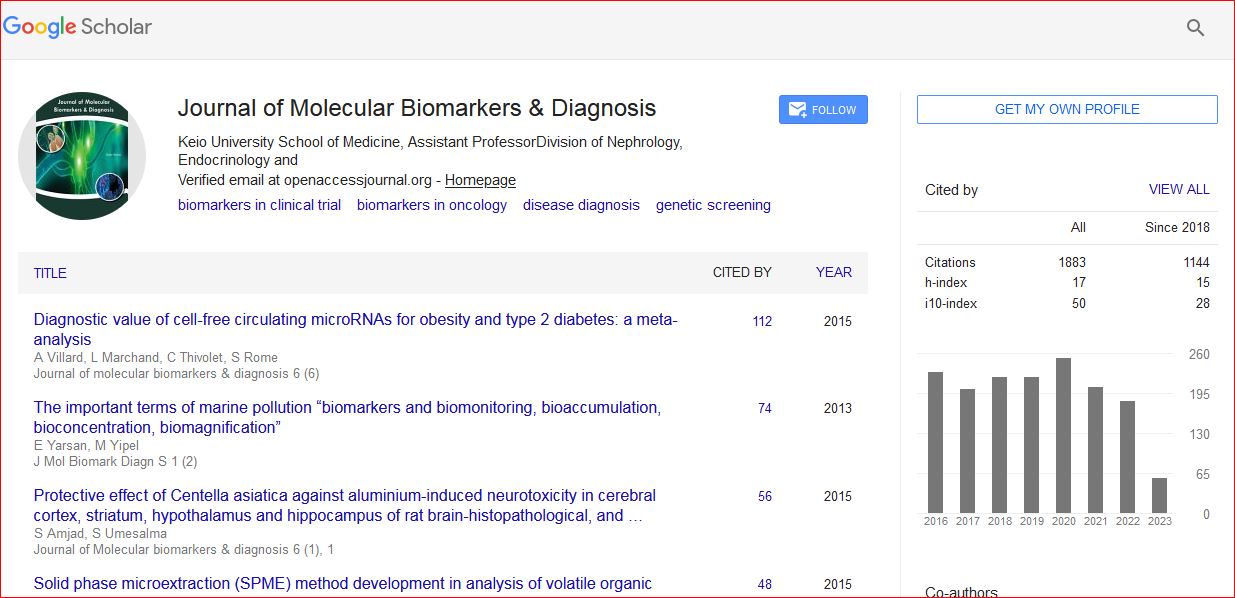
Journal of Molecular Biomarkers & Diagnosis received 2054 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 