Olga Petrovna Sidorova, Bunak M.S, Kotov S.V, Borodataya E.V, Borodin A.V.1 and Polyakov A.V.
M F Vladimirsky Moscow Regional Research and Clinical Institute, Russia Medical Genetics Research Center, Russia
Posters & Accepted Abstracts: J Neurol Disord 2020
Objective: To study changes in the brain and muscles, mitochondrial disorders in patients with myotonic dystrophy. Material and Methods: 12 patients with myotonic dystrophy were examined. Of these, 1 was diagnosed with type 2 and the remaining 1 type of the disease. The diagnosis is confirmed by DNA diagnostics. MRI of the brain was performed. The MRI of lower and upper limb muscles was done on the scanner built around a 1.5 Tesla (T) magnet (Optima MR450w, GE Healthcare) using a T1- and T2-weighted images. For the muscle damage quantification by using MRI a qualitative muscle grading scale, developed by Mercuri et al., (2002) is used. Muscle atrophy grade evaluated during MRI can be divided according to the following criteria: Grade 1 – Early moth-eaten appearance with scattered small areas of increased signal; Grade 2a – Late moth-eaten appearance with numerous discrete areas of increased signal with beginning confluence, comprising less than 30% of the volume of the individual muscle; Grade 2b–Late moth-eaten appearance with numerous discrete areas of increased signal with beginning confluence, comprising 30–60% of the volume of the individual muscle; Grade 3–Washed-out appearance, fuzzy appearance due to confluent areas of increased signal; Grade 4–End stage appearance, muscle replaced by increased density of connective tissue and fat, with only a rim of fascia and neurovascular tissue distinguishable. For a quantitative cytochemical study of the activity of mitochondrial enzymes in peripheral blood lymphocytes, the method proposed by A.G.E. Pearse in the modification of P.P. Narcissov. The activity of four mitochondrial enzymes being involved in carbohydrate (lactate dehydrogenase, LDH), amino acid (glutamate dehydrogenase, GLDH), and fat (α-glycerophosphate dehydrogenase, α-GPDH) metabolism, as well as the enzyme of complex II of mitochondrial respiratory chain (succinate dehydrogenase, SDH) were evaluated. We also measured blood lactate level before and after a carbohydrate loading. Results: MRI of the limb muscles was performed in 6 patients with myotonic dystrophy. In all patients, the medial head of the gastrocnemius muscle was affected. From 2 to 4 points. In 5 out of 6 patients, the soleus muscles were involved in the pathological process. In 4 patients, a lesion of the intermediate broad thigh muscle, the medial broad thigh muscle, the anterior tibial muscle, the long extensor of the thumb, the long extensor of the toes of the foot, the long fibular muscle, the short calf muscle and the lateral head of the gastrocnemius muscle were noted. MRI of the brain was investigated in 5 patients. In 4 patients revealed the presence of subcortical foci in the white matter of the brain. The mean values of the activity of mitochondrial enzymes LDH, α-GPDH, GDH and LDH were statistically significantly reduced (p <0.05). The decrease in the activity of mitochondrial enzymes LDH and α-GPDH was in all the examined patients. The activity of these enzymes was within the limits of normal values in one patient and LDH - in 7. Blood lactate before and after meals exceeded normal values (2.2 mmol / l). The median indicator of blood lactate before the meal was 2.8, after the meal - 3.2. The difference is not statistically significant. Conclusions: Thus, in patients with myotonic dystrophy, not only damage to the muscles of the extremities was revealed, but also damage to the white matter of the brain, probably due to pronounced mitochondrial disorders in these patients. The revealed changes are indications for the appointment of energy-tropic therapy - carnitine and carnosine preparations
Olga Petrovna Sidorova Graduated from Russian National Research Medical University named after N.I. Pirogov in Moscow, residency and graduate school in Neurology. She defended her doctoral dissertation in the specialty of Neurology. She works as a Professor in the Department of Neurology of the Faculty of Advanced Medical Doctors of the Moscow Regional Clinical Research Institute. The chief of Neurological Department is Professor Sergey Kotov. Her research interests - Mitochondrial disorders in neurological diseases, hereditary neurological diseases, myasthenia gravis, photopheresis (extracorporeal photochemotherapy) in autoimmune neurological diseases and porphyria in neurology (acute, chronic and latent course). Together with Hanns Lochmuller, a new mutation has been identified in the gene responsible for one of the forms of congenital myasthenic syndrome.Studies mitochondrial disorders using A.G.E. Pearse in patients with various neurological pathologies.
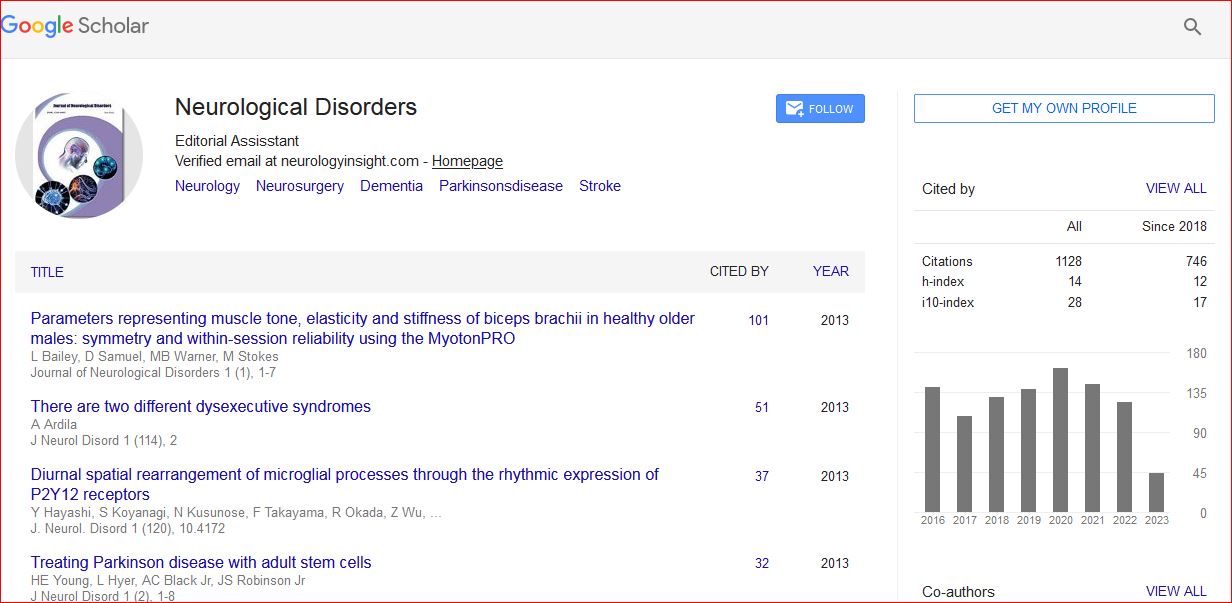
Neurological Disorders received 1343 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 