Istvan Prazsak*, Akos Harangozo, Nikolett Fodor, Balazs Kakuk, Zsolt Csabai, Zsolt Boldogkoi and Dora Tombacz
University of Szeged, Hungary
Posters & Accepted Abstracts: J AIDS Clin Res
In order to understand how viruses are capable of conducting their life cycles and manipulating host immune system it is important to know their gene expression pattern. Existing high-throughput, Short- Read Transcriptome Sequencing Methods (SRS), such as Illumina sequencing have limitations in capturing all possible mRNA isoforms produced by a virus. We circumvent those difficulties by using Long-Read Sequencing (LRS) and found many new, hitherto unknown viral mRNAs in a variety of length among the members of Herpesviruses [1–4]. The Herpesvirus genomes, like other viral genomes consist of complex, gene-dense structures, which can be explained by the parsimonious evolution of viruses, which also enables their high coding capacity. For SRS is difficult, unless it is unable to distinguish these complex transcriptomic structures, however LRS can detect length isoforms of mRNAs very well, especially in overlapping transcriptomic regions [5]. Albeit, many newly found transcript isoforms of Herpesviruses remained unevaluated, due to the low level of abundance, or uncertain detection of transcriptional start and end positions [6,7]. Therefore, the reanalysis of transcriptomic data is in demand. A new integrative elaboration of SRS and LRS transcriptomic analysis of transcriptomic data can resolve the hiatus of Herpesvirus transcriptome annotation. With this new approach, we were able to reconstitute - to our knowledge - the most precisely annotated lytic transcriptome of Varicella Zoster Virus. Results: We have collected transcriptomic data from the NCBI GenBank, which represented all of the uploaded VZV SRS and LRS raw files up to date. Altogether more than 2.5 Billions of reads were used from the results of seven different research groups. We have used Illumina HiSeq, as well as Next Seq CAGE data files and compared them to the downloaded data files of Oxford Nanopore Technologies (ONT) MinION cDNA as well as dRNA sequencing libraries, including Cap-selected, polyA-selected cDNA and dRNA. To enhance the statistical variability of samples for the detection of transcription start (TSS) and end positions (TES) as well as intron positions we re-sequenced the lytic VZV transcriptome. Viruses were propagated in MRC-5 cells and whole transcriptome was sequenced using a random, a targeted and a Terminator treated library preparation protocol on ONT MinION platform. In our bioinformatics pipeline raw reads were treated according their sequencing platform. SRS reads have been trimmed and quality checked by using Trimgalore and FastQC prior to mapping by STAR aligner to the reference genome of VZV (NC_001348.1), thereafter intron-donor/acceptor positions were determined. Furthermore, mapped Illumina reads were re-analyzed by using CAGEfightR to determine TSS positions. Raw LRS data have been re-basecalled by Guppy and mapped by minimap2 to the reference genome of VZV. The LoRTIA tool - developed by our team [8] - was used to access TSS, TES and intron positions from LRS data. Conclusion: By using our integrative analysis, the majority of previously described TSS, TES and intron positions were confirmed and several new, hitherto unknown, intron isoforms and 5’ UTR or 3’ UTR length isoforms of viral transcripts were detected. Our data support the hypothesis, that viral transcription can be analyzed with base-precision by using high coverage SRS, however there is a 2-5 nt uncertainty within 10 nt window within the same sample using different software for TSS determination. We were able to determine at least forty new TSSs, twenty new TESs and thirty new introns in the VZV transcriptome. Importance of Research: We have used a multiplatform and integrative approach of independent transcriptomic data to obtain a detailed transcriptional landscape of transcriptional positions of lytic VZV transcriptome combining both ONT MinION and Illumina sequencing techniques. Solely short read sequencing is impaired to reconstruct overlapping transcript isoforms unlike long read sequencing techniques; however LRS can fail to detect intron isoforms in low abundance. Combining methods, we utilized the advantages of the two different sequencing platforms and revealed a complex mesh of viral RNAs in VZV transcriptome Funding: Istvan Prazsak was supported by UNKP-20-4 -SZTE-140 New National Excellence Program of the Ministry for Innovation and Technology from the source of the National Research, Development and Innovation Fund. Akos Harangozo was supported by the National Academy of Scientist Education (FEIF/646- 4/2021-ITM_SZERZ). This work was also supported by the National Research, Development and Innovation Office K128247 (Zsolt Boldogkoi) and FK128252 (Dora Tombacz).
References :
1. Moldován N, Tombácz D, Szűcs A, et al. Multi-platform sequencing approach reveals a novel transcriptome profile in pseudorabies virus. Front Microbiol. 2018;8:2708.
2. Tombácz D, Csabai Z, Oláh P, et al. Full-length isoform sequencing reveals novel transcripts and substantial transcriptional overlaps in a herpesvirus. PLoS One. 2016;11.
3. Balázs Z, Tombácz D, Szucs A, et al. Long-read sequencing of human cytomegalovirus transcriptome reveals rna isoforms carrying distinct coding potentials. Sci Rep. 2017;7.
4. Tombácz D, Balázs Z, Csabai Z, et al. Long-read sequencing revealed an extensive transcript complexity in herpesviruses. Front Genet. 2018.
5. Boldogk�?i Z, Moldován N, Balázs Z, et al. Long-read sequencing: A powerful tool in viral transcriptome research. Trends Microbiol. 2019.
6. Depledge DP, Puthankalam Srinivas K, Sadaoka T, et al. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen.
7. Prazsák I, Moldován N, Balázs Z, et al. Long-read sequencing uncovers a complex transcriptome topology in varicella zoster virus. BMC Genomics. 2018.
8. Balázs Z. LoRTIA - long-read RNA-seq transcript isoform annotator toolkit. 2019.
Istvan Prazsak, PhD, is an assistant professor at the University of Szeged, Hungary, Department of Medical Biology. He earned MsC in biology and environmental sciences in 2005. He worked as a pre-doctorate researcher at the Department of Genetics, University of Szeged, Hungary, where his research interest covered phylogenetics of arthropod taxa, including Chelicerata. He described a new species of ladybird spiders. From 2010 he joined to the 3G Genomics research group at the Department of Medical Biology, Faculty of Medicine, University of Szeged and focused on the recombinant gene technology and later on transcriptomics of Herpesviruses. From 2018 he has been a PhD student at the Doctoral School of Interdisciplinary Medicine, University of Szeged and Faculty of Medicine under the supervision of Prof Zsolt Boldogkoi and Dr Dora Tombacz. He earned PhD in 2021, entitled: “The Long Read Sequencing of the Varicella-Zoster Virus and Vaccinia Virus Transcriptome”.
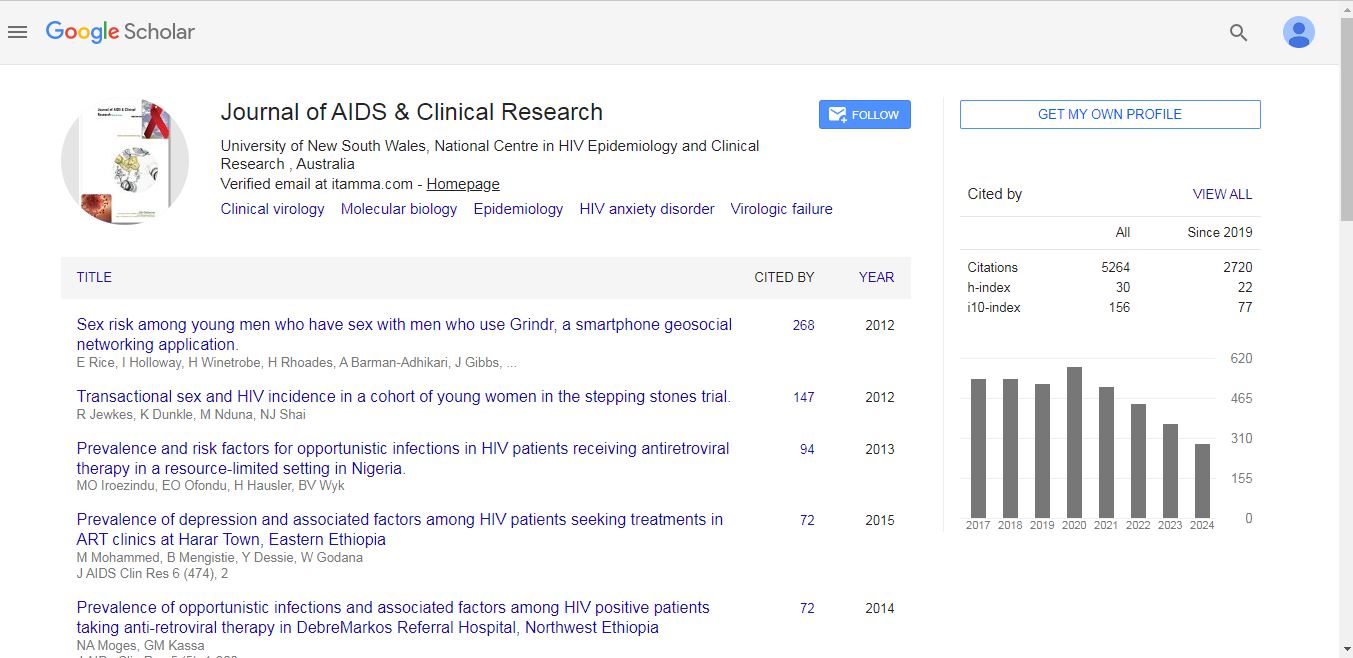
Journal of AIDS & Clinical Research received 5264 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 