Aziza Al-Rafiah
King Abdul Aziza University, Saudi Arabia
Posters & Accepted Abstracts: J Neurol Disord
Spinal muscular atrophy (SMA) is a devastating childhood motor neuron disease caused by mutations in the survival motor neuron 1 gene (SMN1). SMN1 and SMN2 are nearly identical genes producing the survival of motor neuron (SMN) protein. SMN protein plays a crucial role in mRNA splicing and �?²-actin mRNA transport along the axons. In SMA the mutation leads to the loss of SMN1, which cannot be fully compensated by the SMN2 gene, which predominantly produces a truncated protein. The loss or reduction of SMN protein leads to motor axonal defects and motor neuron cell death. There are currently no treatments available but therapies have focused on increasing SMN through replacing SMN1 or increasing full length SMN from SMN2. The actin-binding protein Plastin 3 (PLS3) has been reported as a modifier for SMA, making it a potential therapeutic target. Recently, it was shown that the overexpression of the PLS3 gene improved axonal outgrowth in SMN- deficient motor neurons of SMA Zebrafish and cultured motor neurons from mouse embryos. Gene therapy using viral vectors was carried out in vitro and in vivo to assess whether the overexpression of PLS3 could rescue neuronal loss in SMA and be developed as a therapy. The SMN�?�?7 mouse model produces low levels of SMN, modelling severe SMA disease with an average lifespan of 12 days and loss of motor neurons. This study has established that the SMN�?�?7 mice have little or no detectable PLS3 from birth, making it a good model for developing PLS3 gene therapy. Lentiviral vectors were able to upregulate PLS3 expression in different cell lines. Transduction of NSC34 cells with LV-PLS3 vector led to a five-fold increase in expression of PLS3 compared to controls. In smn-deficient MNs, expression of PLS3 restored axonal length and showed a strong neuroprotective effect. Pre-clinical in vivo proof-of-concept studies using adeno-associated virus serotype 9 (AAV9) encoding PLS3 in SMN�?�?7 mice showed high transduction efficiency and overexpression of PLS3 specifically targeted to neurons in the central nervous system (CNS). This led to a small but significant increase of lifespan by 54%. However, PLS3 was not able to prevent disease onset. Although there was no improvement of phenotype, this study has demonstrated the potential use of PLS3 as a target for gene therapy, possibly in conjunction with other modulators of disease.
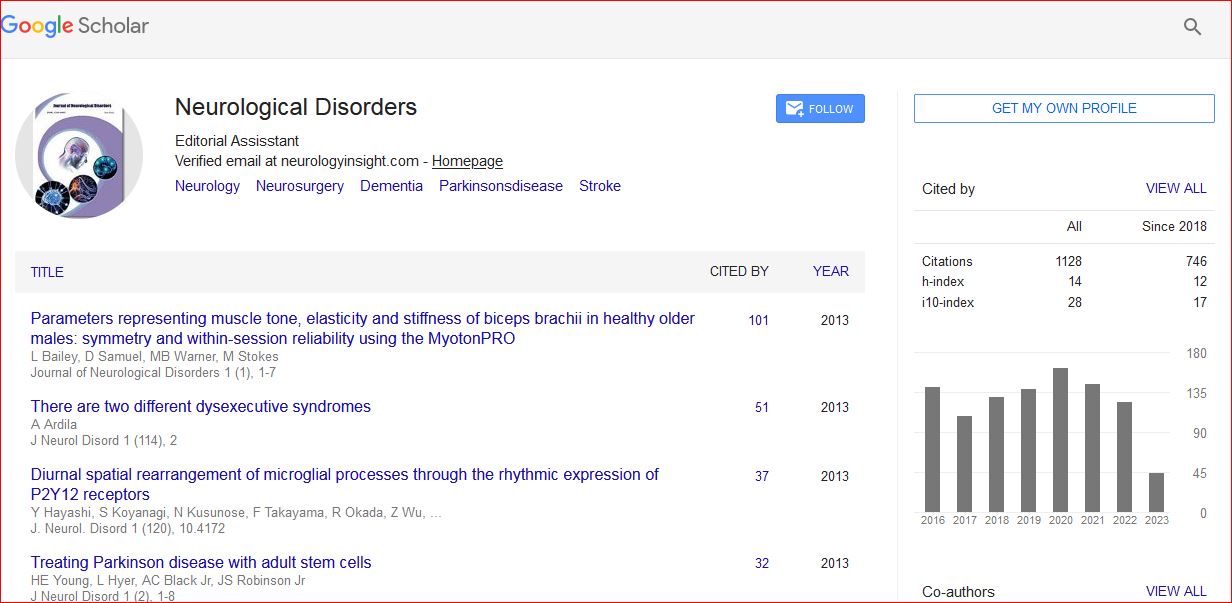
Neurological Disorders received 1343 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 