John K Healy
Sydney University, Australia
Posters-Accepted Abstracts: J Nephrol Ther
Investigation of PHAII has led to a marked expansion of knowledge of distal renal tubular electrolyte handling, which controls critical final urinary composition. This disease, found from infancy to late adulthood, is characterized by hyperkalemia, with up to 9 mmol/l plasma K+, despite a normal glomerular filtration rate. The hyperkalemis leads to acidosis, sometimes severe enough to limit growth. Hypercalciuria and renal calculi may occur. Hypertension occurs in 75% of patients. Plasma renin is depressed and plasma aldosterone is low normal. Even if normotensive at presentation, hypertension generally develops over about 30 years. The hypertension is linked to increased Na+ and Cl- reabsorption through the Na+-Cl-co-transporter (NCC) in the distal convoluted tubule (DCT). Although frequently familial, 36% of cases of PHAII are de novo. The hyperkalemia has been ascribed in humans to (a) suppression of renal outer medullary K+ secretion (ROMK), or (b) increased chloride (Cl-) reabsorption (a Cl- shunt) depressing the degree of negativity at the site of K+ secretion in exchange for Na+ at the epithelial Na+ channels (ENaC), or (c) decreased Na+ delivery to the ENaC in the late distal nephron reducing K= secretion at that site. In 2000, geneticists discovered a serine-threonine kinase in rats they called WNK1, because it lacked lysine at the usual catalytic point (with no lysine (=K)), and soon WNK��?s 2,3 and 4 were found. In man, mutations of WNK��?s 1 and 4 (WNK��?s 1 and 4) localised to the distal nephron in the critical site of electrolyte homeostasis, and were found to cause PHAII by failing to cause suppression of the NCC, which is the function of normal WNK��?s. It was eventually found that mutations in WNK1 and WNK4 cause 13% of PHAII. Recently a further system regulating distal electrolyte handling called cullin 3 and kelch-like3 was found in an E3 ubiquitination ligase complex in the distal nephron, and mutations of these (CUL3 and KLHL3) cause 79% of PHAII. Clinical severity of PHAII is, in decreasing order, CUL3, recessive KLHL3, dominant KLHL3, WNK4, andWNK1. CUL3 and KLHL3 block ubiquitination (removal for degradation) of WNK4, accumulation of which causes suppression of ROMK and so can cause hyperkalemia as in mechanism (a) above. Mutant WNK4 also stimulates Cl- reabsorption by the paracellular pathway, supportive of (b) above. Although over-activity of the NCC may initially decrease Na+ delivery to the ENaC as in (c) above, in a stable state Na+ delivery to the ENaC would be normal, and sodium sulphate and sodium bicarbonate infusion studies, with ample delivery of Na+ to the ENaC, failed to provide even a quantitatively normal kaliuretic response, weakening the argument for (c) above. Thiazides are the most effective treatment of PHAII, as they inhibit the NCC. They produce mild hypovolemia in regular use, which may cause antidiuretic hormone release and this is a potent stimulator of K+ secretion. Thus, dDAVP rapidly reverses the hyperkalemia of PHAII, and this has been used successfully in treatment of this condition. Na+ restriction should be used with caution as it has been shown to increase plasma K+ in some studies, while in others it has reduced it or had no effect. If still needed, dietary K+ restriction may be effective.
Email: jkhealy@healyblore.com
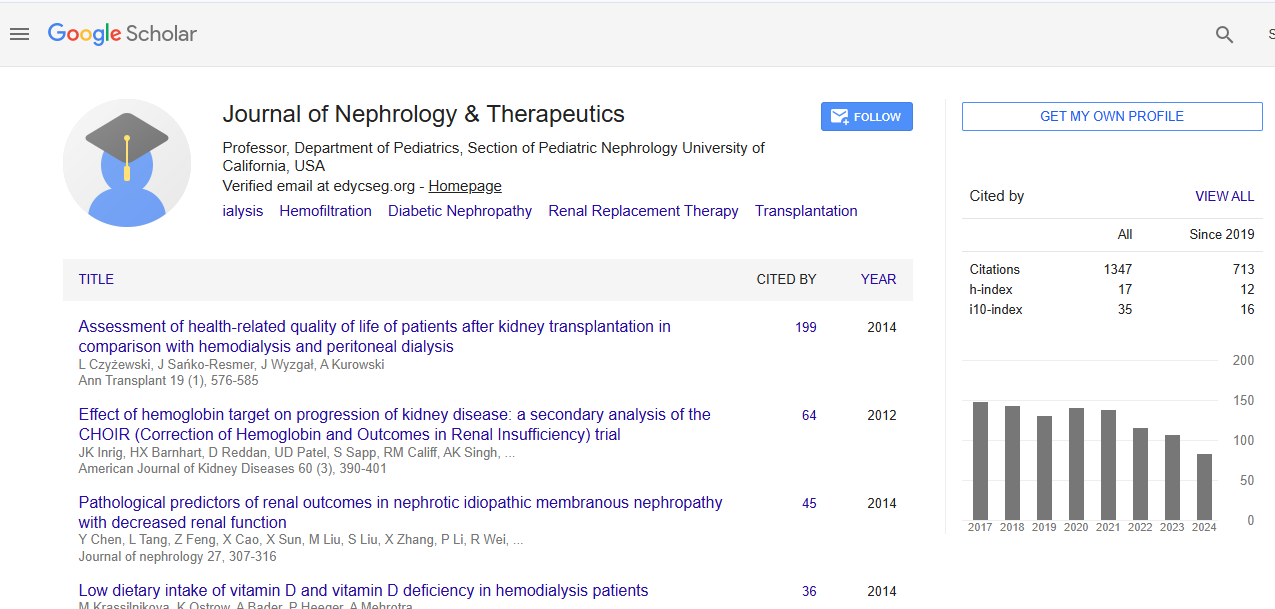
Journal of Nephrology & Therapeutics received 784 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 