Majdy Idrees
Posters-Accepted Abstracts: J Pulm Respir Med
Pulmonary arterial hypertension (PAH) is a disease of the small pulmonary arteries characterized by vascular obstruction leading to progressive elevations in pulmonary vascular resistance and pulmonary artery pressures and ultimately right heart failure. In PAH, a distinct vascular lesion known as the plexiform lesion is considered the histological hallmark of this disease. Although plexiform lesions involve the three components: Smooth muscle layer, adventitia and the endothelium, most studies have focused on the smooth muscle component and the role of vasodilators although less than 10% of PAH patients respond fully to simple vasodilators therapy. Thus, the development of new theories has become necessary. Subsequently, augmented endothelial cell proliferation leading to complicated capillary-like channels (angio-proliferation) as the main component of the plexiform lesion has been reported. Furthermore, the discovery of endothelial monoclonality in plexiform lesions of idiopathic PAH further support the �??neoplastic hypothesis�?� of the disease. Several mutations have been linked to both angio-proliferation and inhibition of apoptosis in endothelial cells from plexiform lesions, the whole mark of tumor formation. For instance, alterations in transforming growth factor-β (TGF-β) receptor II may turn endothelial cell insensitive to the cell growth-controlling effects of TGF- β5. Moreover, expression of anti-apoptotic protein survivin has been reported in PAH plexiform lesions. Furthermore, a shift from oxidative phosphorylation to aerobic glycolysis (the Warburg effect) which is originally described in tumor cells has been also described in PAH. Although the recently published IMPRES study using Imatinib, a tyrosine kinase inhibitor in treating severe pulmonary hypertension was considered negative because of the drug toxicity, it has nevertheless confirmed the significant efficacy of these agents in improving the exercise tolerance, symptoms and hemodynamics and further supports the neoplastic features of the disease. This should certainly open the door to develop new diagnostic techniques and targeted therapies against the vascular remodeling component.
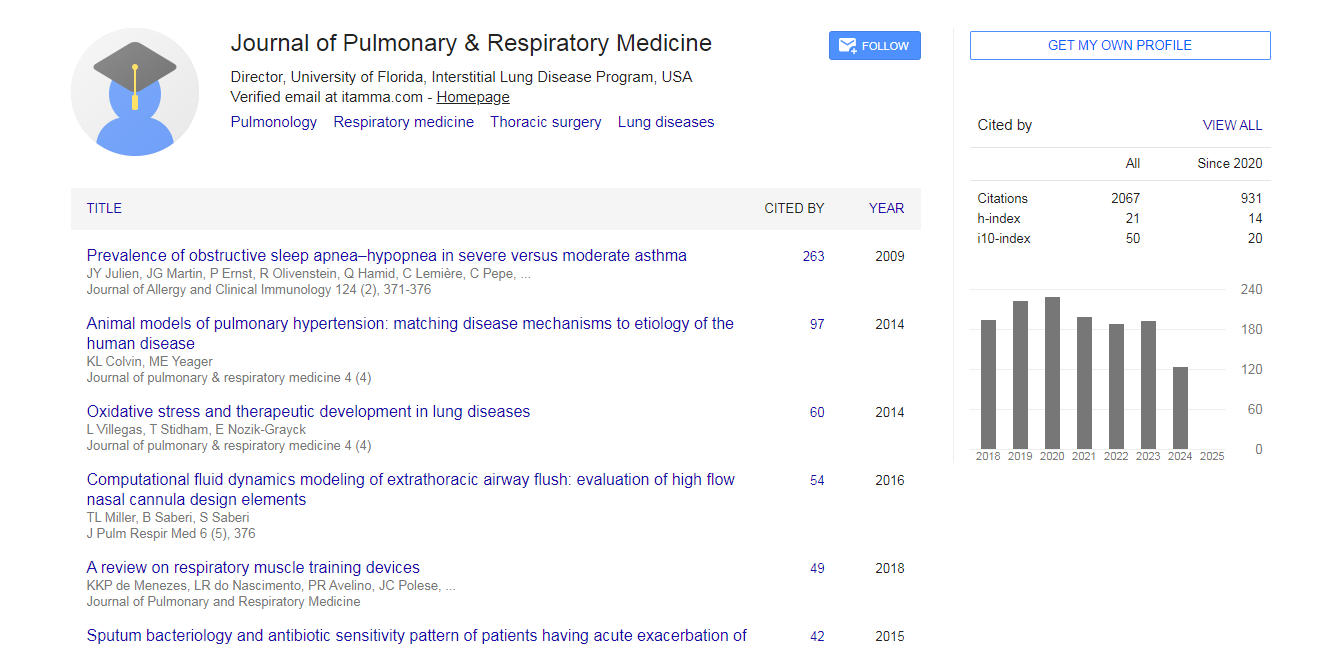
Pulmonary & Respiratory Medicine received 1690 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 