Claudio Nicolini, Marine Bozdaganyan, Philipp Orekhov and Eugenia Pechkova
University of Genoa, Italy
Posters & Accepted Abstracts: J Material Sci Eng
New trends and perspectives in protein nanocrystallography are emerging at the intersection of advances in nanotechnology (Langmuir-Blodgett and Anodic Porous Allumina), functional (microarray, cell free expression and SNAP) and structural synchrotron radiation (third generation sources trillion times more brilliant and XFEL both requiring quite smaller crystals and Monte Carlo simulation) proteomics. It should be noted that nanocrystallography here does not refer to crystals of nanometer size or to nanodrop crystallization technology, but to the significant applications for crystallography emerging in our labs at the interface of Langmuir-Blodgett engineering, organic chemistry, molecular dynamics and label-free protein arrays, utilizing bacterial hell's gate globin, octopus rhodopsin, bovine cytochrome, human kinase, laccase and many other proteins. Molecular dynamics of proteins in aqueous environments are now routinely performed at an atomic level on the time scale up to several microseconds. MD simulations have been useful for predicting drug binding sites not available in protein X-ray structures, elucidating the origins of drug specificity, computing binding energies for ligand-protein systems and many other applications. Crystallographic methods have played an immense role in providing detailed biomolecular structural information and have been fundamental in the development of our understanding of the structure-function relationship. At the same time, crystallographic models can display an overreliance on static representations of biomolecular structure, despite the fact that biomolecules are intrinsically dynamic. MD simulations help to reveal these dynamics. The Monte Carlo approach has gained wide acceptance in multiple fields of natural science and technology as an effective tool for rapid integration in the highly dimensional spaces. It is extensively utilized in nuclear physics, molecular modeling, elementary particle physics, analysis of the diffraction data, etc. Here, we utilize the MC simulations in order to track the X-ray photons passage through protein crystals and their interactions. This work is an extension of the previous work, and it aims to improve our understanding of the exact organization of crystals, solute and salt orientation around the proteins. MD simulations of crystal lattices are carried out in different solutions as well as at different temperatures in order to understand the role of salts in crystal stability. In this research, we are planning to compare dynamics of lysozyme crystals obtained using the classical HD method, the LB method and the SG crystals. At the same time using Lomonosov supercomputer, we have developed a computational routine, which allows preparing molecular geometries corresponding to arbitrary protein crystals optimized and sampled using MD simulations and subsequently simulate initial processes of radiational damage in them by means of our Monte Carlo approach.
Email: clannicolini@gmail.com
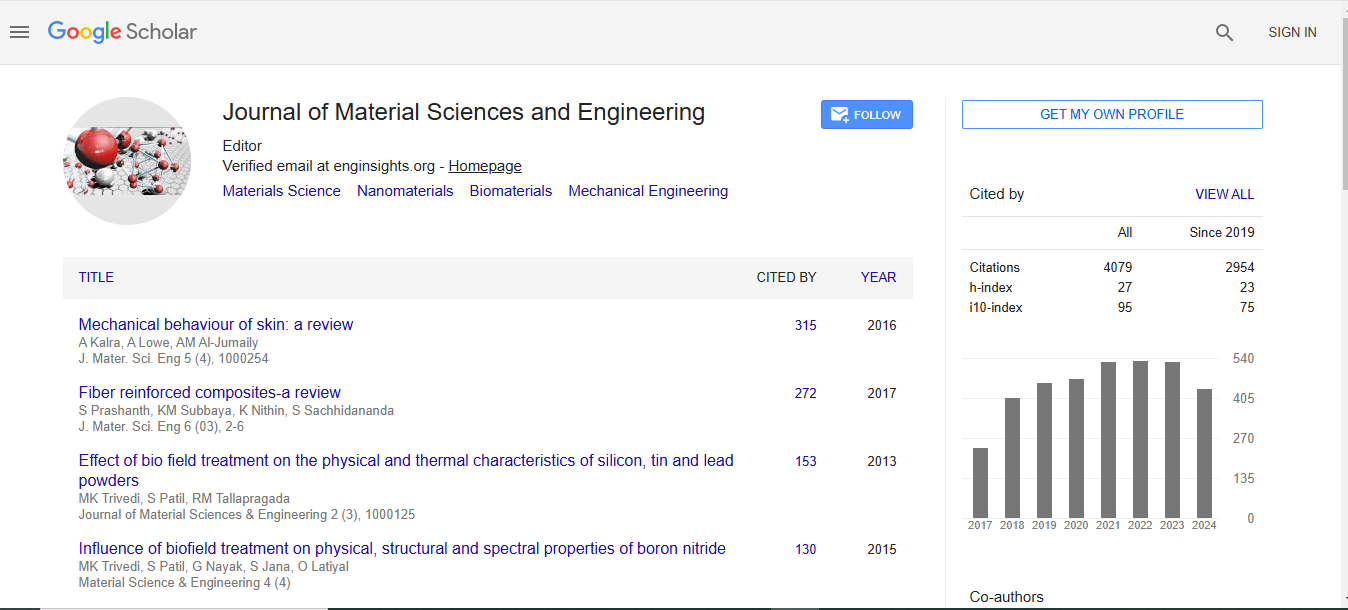
Journal of Material Sciences & Engineering received 3677 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 