Tamilselvi S and Ankanagari Srinivas
Accepted Abstracts: J Bioanal Biomed
Biopharmaceutical drugs have become an essential part of modern pharmacotherapy. These comprise proteins derived from recombinant DNA technology and hybridoma technique. Examples include biological proteins (cytokines, hormones, and clotting factors), monoclonal antibodies, vaccines and cell and tissue based therapies. Living organisms such as plant and animal cells, bacteria, viruses and yeast are employed for the production of biopharmaceuticals. These are developed after patent expiration of innovator biopharmaceuticals and are submitted for separate marketing approval. New drug substance to be introduced for the first time in the country, including vaccines & r-DNA derived products needs to be routed through New Drug Advisory Committee. This also applies to global CTs, and new fixed dose combinations to be introduced for the first time. These applications will be first scrutinized by DCGI office and within the time frame specified and referred to individual New Drug Advisory Committee members of the applicable therapeutic category. Recently Indian government has taken several initiatives towards streamlining the way biosimilars will be regulated thereby showcasing India as a key player in the biosimilar segment. The European Union (EU) has been the first to establish a regulatory framework for marketing authorization application (MAA) and has named these products biosimilars, a term also used by the US FDA. Unlike the conventional, more common small molecular weight human medicines, chemical generics and protein-based medicines exhibit higher molecular weight, complexity in structure and function that can be affected by changes in the manufacturing process. Biologically derived drugs have a much greater potential than chemically synthesized drugs to elicit immune responses. These responses can be rare, sometimes too rare to detect even with any reasonable premarket safety database, and the severity of response can vary considerably. Therefore, biosimilars represent a relatively heterogeneous class of medicinal products and make its regulation quite complicated and challenging in terms of Quality, safety and efficacy when compared to the innovator product. According to the European guideline, a biosimilar is a duplicate of an already approved biopharmaceutical product with similar biologic activity, physicochemical characteristics, efficacy, and safety, based on a full comparability exercise at quality, preclinical and clinical relevant to immunogenicity potential to ensure similar efficacy and safety. Guidance has been given by various Committees for Medicinal Products for Human Use (CHMP) as well as individual scientific advice requested from the European Medicines Agency (EMA) by various companies for the development and regulation of biosimilars. Thus current status of regulation of biosimilars in the EU as well as on future challenges lying ahead for the improvement of the requirements needed for the marketing authorization of biosimilars. Emphasis is given on the quality requirements concerning these medicinal products (biologics). Production of these new products is expected to meet worldwide demand, promote market competition, maintain the incentives for innovation, and sustain the healthcare systems. The licensing of these products however relies on the experience gained with the original biopharmaceuticals.
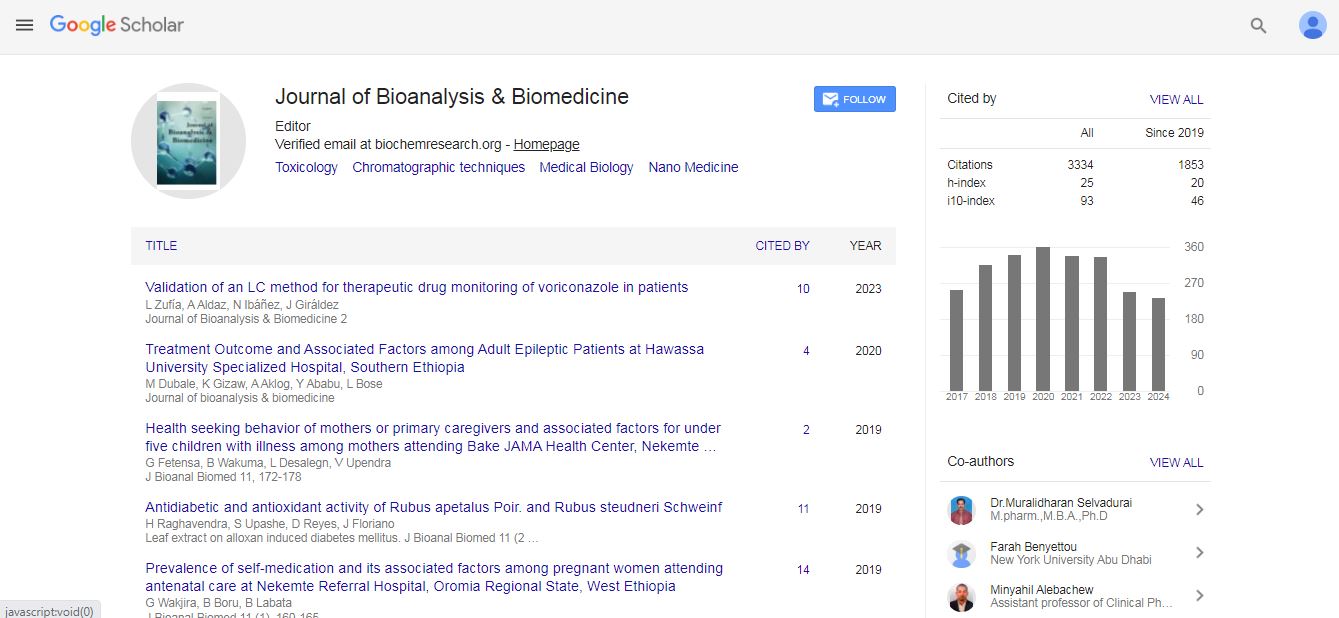
Journal of Bioanalysis & Biomedicine received 3099 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 