Andrey Lisitsa, Olga Kiseleva, Sergei Moshkovskii and Alexander Archakov
Accepted Abstracts: J Mol Biomark Diagn
O ne of the main features of MS based quantification is clinical application used to identify biomarkers associated with diseases. Despite early expectations, proteomics failed to discover the expression biomarkers, withstanding the validation on large cohorts. Putative escape was enlightened by proteoformics, the offspring of proteomics for studying the variety of protein species, called proteoforms and arised as a consequence of single amino acid change, alternative splicing, post- translational modifications, and other yet unexplored molecular events. The phenomenon of SAPs was extensively studied by bottom-up shotgun mass-spectrometry. The new step is to match the MS/MS spectra against the somatic mutations, which can be expected from non-synonymic SNPs of cancer genome. Development of SAP-based digital biomarkers requires a number of clinically-relevant experiments matched against the cancer exomes deciphered by genome sequencing. The pipeline for investigation the information about cancer-associated SAP-based biomarkers is following. The cancer genomes upcoming from the large-scale COSMIC database are filtered to derive the nsSNPs, which can be additionally ranked for their influence onto the protein structure or functional significance. Selected nsSNPs are in silico translated into the protein sequence database. The database is searched towards the MS/MS spectra repositories, either general ones, like PRIDE, or preferably those, convicted to cancer tissue or cell lines. The MS/MS datasets are matched to the peptide?s mutations expected from the cancer genome, withdrawing the unaware cases of modifications while simple preparation. The remaining SAP-containing peptides are investigated for their occurrence in the control samples, and discarded in case of match. The rest has to be validated using the SRM targeted approach, which includes the synthesis of stable isotope standard (SIS) peptides, assay development and optimization. The additional verification has to be performed by PCR to confirm the corresponding mutation at the genome level. Cancer driver genes APC, TP53 и KRAS are affected in 42-71% of cases. However, each individual nsSNP is rarely observed. It means that each tumor accumulates a unique set of spontaneous mutations. There are many mutations to break the proto-oncogene or tumor suppressor; however, the mutation can occur in different sites along the gene. That property of the cancer tumor represents bad news for biomarkers which may reach the 100% specificity, but at the 9% sensitivity, as only 9% of tumors carry the particular mutation. We conclude that despite the technical level is ready to deliver information about proteoform-based biomarkers, still further increase in sensitivity and selectivity is required to validate the concept. The effort can be undertaken in frames of the Chromosome-centric Human Proteome Project, to open up a new format of diagnostics for clinical proteomics.
Andrey Lisitsa graduated from Russian National Research Medical University specializing in biochemistry in 1999 and received his Ph.D. degree in biochemistry in 2002. Awarded from the Government of Russian Federation for the achievements in science and technologies in 2005. In 2007 has got his Doctoral degree in Bioinformatics for development of knowledgebase in the field of Experimental and Bioinformatics Studies of Cytochrome P450 Superfamily. Currently, he is a Head of the Bioinformatics Technologies Laboratory and Deputy Director of Science of the Institute of Biomedical Chemistry, Moscow. Andrey Lisitsa is corresponding member of the Russian Academy of Medical Sciences. His research interests lie in development of new technologies offering unique features for ultra-low level detection of proteins in biosamples.
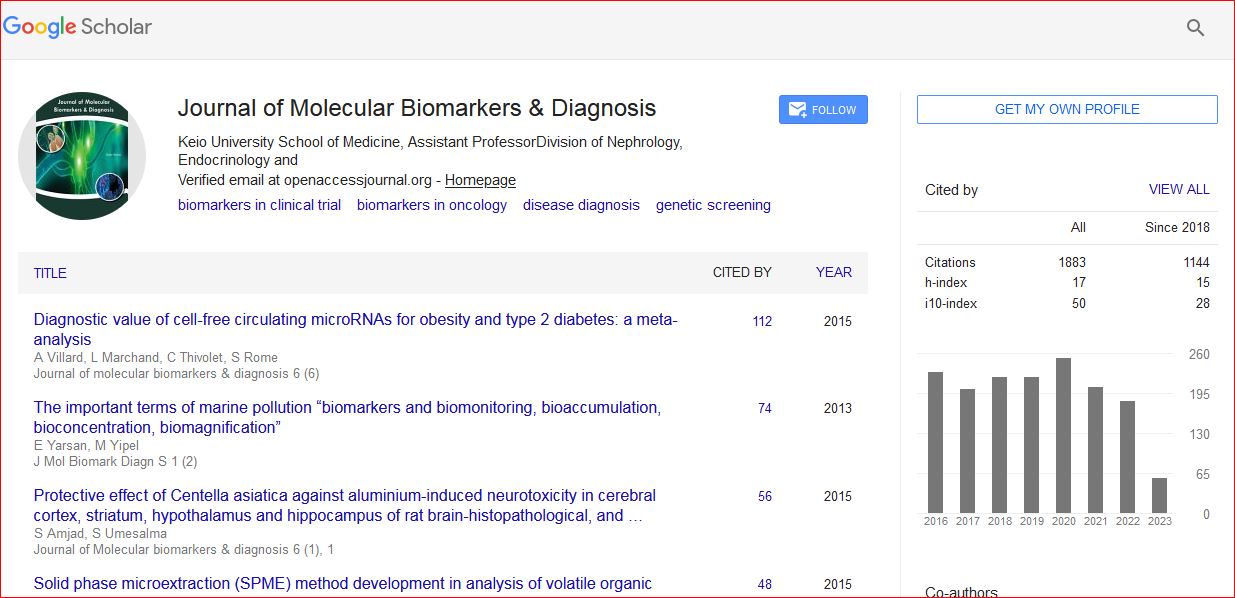
Journal of Molecular Biomarkers & Diagnosis received 2054 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 